- Details
- ICNA
- News
- Hits: 722
 A Phase I clinical trial led by investigators from the University of California, San Francisco (UCSF) has shown that neural stem cells successfully engrafted into the brains of patients and appear to have produced myelin. The findings have been published in the Oct 10, 2012 issue of Science Translational Medicine.
A Phase I clinical trial led by investigators from the University of California, San Francisco (UCSF) has shown that neural stem cells successfully engrafted into the brains of patients and appear to have produced myelin. The findings have been published in the Oct 10, 2012 issue of Science Translational Medicine.
Once transplanted and engrafted, neural stem cells have the potential to differentiate into a number of different brain cell types, depending on the area of the brain into which they are inserted. The sites chosen for the Phase I study were known from animal studies to be the most likely to result in the formation of oligodendrocytes.
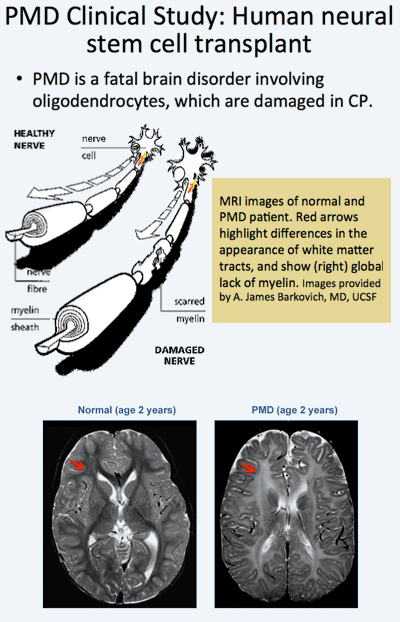 In the trial, allogeneic human neural stem cells (HuCNS-SCs) developed by Stem Cells, Inc., of Newark, California, were injected directly into the frontal lobe white matter of four young children with an early-onset, fatal form of a condition known as Pelizaeus-Merzbacher disease (PMD).
In the trial, allogeneic human neural stem cells (HuCNS-SCs) developed by Stem Cells, Inc., of Newark, California, were injected directly into the frontal lobe white matter of four young children with an early-onset, fatal form of a condition known as Pelizaeus-Merzbacher disease (PMD).
Immunosuppression was administered for 9 months. During 2010-2011, the children with PMD, who were included in the trial, underwent serial neurological evaluations, developmental assessments, and cranial magnetic resonance imaging (MRI) and MR spectroscopy, including high-angular resolution diffusion tensor imaging (DTI). The investigators found evidence that the stem cells had successfully engrafted, receiving blood and nutrients from the surrounding tissue and integrating into the brain.
The Phase 1 trials designed to test safety and preliminary efficacy demonstrated that the neural stem cells were safe in the patients’ brains one year post transplant. No clinical or radiological adverse effects were directly attributed to the donor cells. Reduced T1 and T2 relaxation times were observed in the regions of transplantation 9 months after the procedure in the three subjects. Normalized DTI showed increasing fractional anisotropy and reduced radial diffusivity, consistent with myelination, in the region of transplantation compared to control white matter regions remote to the transplant sites. The MRI findings suggested myelination in the regions that have been transplanted providing indirect evidence that the stem cells had become oligodendrocytes and were producing myelin.
 These results have thus provided for the first time evidence that transplanted neural stem cells are able to produce new myelin in patients with a severe myelination disease. The MRI findings from the clinical trial are further supported by findings from a separate study2 by in a separate study by researchers at Oregon Health & Science University's Papé Family Pediatric Research Institute which showed that neural stem cells injected into mouse models became oligodendrocytes and formed myelin.
These results have thus provided for the first time evidence that transplanted neural stem cells are able to produce new myelin in patients with a severe myelination disease. The MRI findings from the clinical trial are further supported by findings from a separate study2 by in a separate study by researchers at Oregon Health & Science University's Papé Family Pediatric Research Institute which showed that neural stem cells injected into mouse models became oligodendrocytes and formed myelin.
 The PMD clinical trial researchers included Principal investigator David H. Rowitch, MD, PhD, a professor of pediatrics and neurological surgery at UCSF, chief of neonatology at UCSF Benioff Children's Hospital and a Howard Hughes Medical Institute Investigator and co-principal investigator Nalin Gupta, MD, PhD, associate professor of neurological surgery and pediatrics and chief of pediatric neurological surgery at UCSF Benioff Children's Hospital.
The PMD clinical trial researchers included Principal investigator David H. Rowitch, MD, PhD, a professor of pediatrics and neurological surgery at UCSF, chief of neonatology at UCSF Benioff Children's Hospital and a Howard Hughes Medical Institute Investigator and co-principal investigator Nalin Gupta, MD, PhD, associate professor of neurological surgery and pediatrics and chief of pediatric neurological surgery at UCSF Benioff Children's Hospital.
The study, one of the first neural stem cell trials ever conducted in the United States, is symbolical of UCSF’s pioneering role in the stem cell field.
![]() In 1981, Gail Martin, PhD, professor of anatomy, co-discovered embryonic stem cells in mice.
In 1981, Gail Martin, PhD, professor of anatomy, co-discovered embryonic stem cells in mice.
![]() In 2001, Roger Pedersen, PhD, professor emeritus of obstetrics, gynaecology and reproductive sciences, derived two of the first human embryonic stem cell lines.
In 2001, Roger Pedersen, PhD, professor emeritus of obstetrics, gynaecology and reproductive sciences, derived two of the first human embryonic stem cell lines.
![]() In 2012, Shinya Yamanaka, MD, PhD, of the UCSF-affiliated Gladstone Institutes and Kyoto University, received the Nobel Prize in Physiology or Medicine for his discovery that adult cells can be reprogrammed to behave like embryonic stem cells
In 2012, Shinya Yamanaka, MD, PhD, of the UCSF-affiliated Gladstone Institutes and Kyoto University, received the Nobel Prize in Physiology or Medicine for his discovery that adult cells can be reprogrammed to behave like embryonic stem cells
Citations
1Gupta N, Henry RG, Strober J, Kang SM, Lim DA, Bucci M et al. (2012) Neural stem cell engraftment and myelination in the human brain. Sci Transl Med 4 (155):155ra137. DOI: 10.1126/scitranslmed.3004373 PMID: 23052294.
2Uchida N, Chen K, Dohse M, Hansen KD, Dean J, Buser JR et al. (2012) Human neural stem cells induce functional myelination in mice with severe dysmyelination. Sci Transl Med 4 (155):155ra136. DOI: 10.1126/scitranslmed.3004371 PMID: 23052293.
About Pelizaeus-Merzbacher disease (PMD)
- Rare congenital X-linked recessive leukodystrophy
- Incidence of 1:200,000 to 1:500,000
- Caused by mutation of myelin protein proteolipid protein 1 (PLP1), resulting in hypomyelination
- Leading to death between ages 10 and 15
- Oligodendrocytes are unable to myelinate axons, resulting in loss of normal axonal conduction and neurological dysfunction in the short term, eventually leading to axonopathy and neurodegeneration.
- PMD is one of a spectrum of diseases associated with PLP1, which also includes Spastic Paraplegia Type 2 (SPG2)
- Four types recognised
- Congenital PMD -early-onset severe form of PMD presents with profound neurodevelopmental deficits.
- Classic PMD, in which the early symptoms include muscle weakness, involuntary movements of the eyes (nystagmus), and delays in motor development within the first year of life;
- Complicated SPG2, which features motor development issues and brain involvement, and,
- Pure SPG2, which includes cases of PMD that do not have neurologic complications.
Further Reading
Pelizaeus-Merzbacher disease (PMD)
Read More
- Details
- ICNA
- News
- Hits: 598
 The ICNA is happy to announce an educational program to be held in Pune, India on January 19-20, 2013 in collaboration with Pune Neurological Society.
The ICNA is happy to announce an educational program to be held in Pune, India on January 19-20, 2013 in collaboration with Pune Neurological Society.
The ICNA will be represented by Doctors Harry Chugani, Banu Anlar, Linda De Meirleir, Peter Camfield, Carol Camfield.
The local organizer is Dr Nandan Yardi, K.E.M. Hospital Research Centre, Pune
Read More
- Details
- ICNA
- News
- Hits: 834
The 48-week results from the ongoing Phase IIb clinical trial of eteplirsen for the treatment of Duchenne muscular dystrophy (DMD)has been announced. Eteplirsen is an exon-skipping compound that addresses one of the underlying genetic defects in Duchenne muscular dystrophy.
At 48 weeks eteplirsen demonstrated a significant and unprecedented clinical benefit on the primary clinical outcome measure, the 6-minute walk test, and met the primary efficacy endpoint of the study, an increase in novel dystrophin. The results represent the potential medical breakthrough that eteplirsen represents for the treatment of DMD.
Although only about 13% of boys with Duchenne muscular dystrophy have the specific mutation targeted by eteplirsen, the implications for all Duchenne muscular dystrophy patients with related genetic mutations are clearly evident.
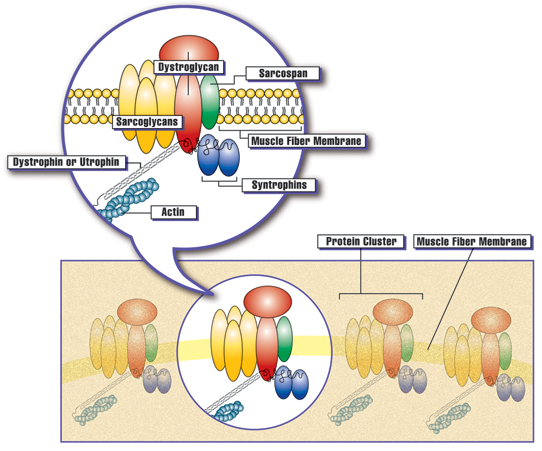 Clusters of proteins are spaced at intervals along the membrane of each muscle fiber. The presence of dystrophin, or the related protein utrophin, appears to be necessary for the other proteins in the cluster to assemble at the membrane. Courtesy: Muscular Dystrophy Association
Clusters of proteins are spaced at intervals along the membrane of each muscle fiber. The presence of dystrophin, or the related protein utrophin, appears to be necessary for the other proteins in the cluster to assemble at the membrane. Courtesy: Muscular Dystrophy Association
Trial Results
Eteplirsen administered once weekly at either 30mg/kg or 50mg/kg for 48 weeks (n=8) resulted in a statistically significant increase in dystrophin-positive fibers of 47.0% of normal.
The placebo/delayed treatment cohort, which received 24 weeks of eteplirsen at either 30mg/kg or 50mg/kg following 24 weeks of placebo (n=4), produced a statistically significant increase in dystrophin-positive fibers of 38.3% of normal.
In addition, eteplirsen administered once weekly at 50mg/kg over 48 weeks resulted in a 89.4 meter benefit compared to patients in our placebo/delayed treatment arm, those patients who received placebo for 24 weeks followed by 24 weeks of treatment with eteplirsen in the open-label extension of the study.
Upon evaluating all study participants through 48 weeks, no treatment-related adverse events which further demonstrate the highly favorable safety profile of eteplirsen.
An abstract describing the results from this Phase IIb extension study has been accepted as part of the World Muscle Society (WMS) Congress's Late-Breaking Science program in Perth, Australia during October 9 to October 13, 2012.
Principal investigator, Jerry R. Mendell, M.D. of Nationwide Children's Hospital, will present the data "Results at 48 Weeks of a Phase IIb Extension Study of the Exon-Skipping Drug Eteplirsen in Patients with Duchenne muscular dystrophy (DMD)" at the World Muscle Society (WMS) Congress's Late-Breaking Science program in Perth, Australia on October 13 at 4:00 p.m. WST UTC +8 hours/4:00 a.m. EDT.
Summary of Dystrophin: Eteplirsen-Treated Patients in All Dose Groups through Week 48*
| Treatment Arm | Mean Change from Baseline in % Dystrophin-Positive Fibers | p-value | |||
| Eterplirsen (both doses): 48 wks of Tx (n=8) | 47.0 | ≤0.001 | |||
| Eteplirsen 50 mg/kg (n=4) | 41.7 | ≤0.008 | |||
| Eteplirsen 30 mg/kg (n=4) | 52.1 | ≤0.001 | |||
| Placebo/Delayed Tx: 24 wks of Tx (n=4) | 38.3 | ≤0.009 | |||
| Placebo/50 mg/kg Delayed-Tx (n=2) | 42.9 | ns | |||
| Placebo/30 mg/kg Delayed-Tx (n=2) | 34.2 | ns | |||
* Values based on Immunofluorescence using anti-dystrophin antibody MANDYS106
Modified Intent-to-Treat (mITT)
The 6MWT results were further analyzed using the mITT population which excluded two patients who were randomized to the 30 mg/kg weekly eteplirsen cohort who showed signs of rapid disease progression within weeks after enrollment and were unable to perform measures of ambulation beyond 24 weeks. This mITT population consisted of 10 patients (4 eteplirsen-treated patients receiving 50 mg/kg weekly, 2 eteplirsen-treated patients receiving 30 mg/kg weekly, and 4 placebo/delayed-treatment patients).
Summary of 6MWT: Eteplirsen versus Placebo/Delayed-Treatment to Week 48*
| Treatment Arm | Mean Change from Baseline in 6MWT (meters) | Estimated Treatment Effect (Eteplirsen minus Placebo/Delayed-Tx) | p-value | |||
| Placebo/Delayed-Tx (n=4) | -60.3 | |||||
| Eteplirsen 50 mg/kg (n=4) | +27.1 | 87.4 m | ≤0.001 | |||
| Eteplirsen Both Doses (n=6) | +7.3 | 67.3 m | ≤0.001 | |||
| Eteplirsen 30 mg/kg (n=2) | -31.5 | 28.8 m | ns | |||
*Note: Analysis based on Mixed Model Repeated Measures test
Summary of Additional Sub-Group Analyses at Week 48*
| Subset | Mean 6MWT Change from Baseline (meters) | Estimated Treatment Benefit (Eteplirsen minus Placebo/delayed-Tx) | p-value | |||
| Placebo/delayed Tx: < 9.5 yrs at baseline (n=2; mean=7.6 yrs) |
-42.3 | 58.9 m | ≤0.038 | |||
| Eteplirsen: < 9.5 yrs at baseline (n=3; mean=8.4 yrs) |
+16.5 | |||||
| Placebo/delayed Tx: ≥9.5 yrs at baseline (n=2; mean=10.1 yrs) |
-63.5 | 52.1 m | ns | |||
| Eteplirsen: ≥9.5 yrs at baseline (n=3; mean=10.4 yrs) |
-11.3 | |||||
| Placebo/delayed Tx: Higher 6MWT baseline (n=2; mean=422m) |
-53.5 | 93.8 m | ≤0.001 | |||
| Eteplirsen: Higher 6MWT baseline (n=3; mean=424m) |
+40.3 | |||||
| Placebo/delayed Tx: Lower 6MWT baseline (n=2; mean=367m) |
-65.8 | 39.6 m | ns | |||
| Eteplirsen: Lower 6MWT baseline (n=3; mean=375m) |
-26.2 | |||||
| Placebo/delayed Tx: Genotype 49-50 deletion (n=3; age mean=9.2 yrs, 6MWT BL mean=397m) |
-69.0 | 83.4 m | ≤0.001 | |||
| Eteplirsen: Genotype 49-50 deletion (n=2; age mean=9.1 yrs, 6MWT BL mean=383m) |
+14.4 | |||||
* Note: Analysis based on Mixed Model Repeated Measures test
About Study 201 and Study 202 (Phase IIb Eteplirsen Study)
Study 4658-US-201 was conducted at Nationwide Children's Hospital in Columbus, Ohio. Twelve boys meeting the inclusion criteria being between 7 and 13 years of age with appropriate deletions of the dystrophin gene that confirm eligibility for treatment with an exon-51 skipping drug, received double-blind IV infusions of placebo (n=4), 30 mg/kg of eteplirsen (n=4), or 50 mg/kg of eteplirsen once weekly for 24 weeks (n=4). Muscle biopsies for evaluation of dystrophin were obtained at baseline for all subjects, and after 12 weeks for patients in the 50 mg/kg cohort and after 24 weeks for patients in the 30 mg/kg cohort. Two placebo patients were randomized to the 30 mg/kg cohort and two placebo patients were randomized to the 50 mg/kg cohort. This study design allowed to investigate the relationship of dose and duration of eteplirsen treatment on the production of dystrophin over the course of the 24-week study.
Study 4658-US-202 is the extension study to 201 and continues to assess the long-term safety and efficacy of open-label eteplirsen. The four placebo patients were rolled over to open-label eteplirsen at week 24, with six patients on 30 mgs/kg, and six patients on 50 mgs/kg. Third biopsies occurred at 48 weeks in the original study 201 treated patients, and at 24 weeks, the same time point, in the original placebo patients. 6MWT was performed at 32 weeks, 36 weeks, 48 weeks and will continue to be performed every 12 weeks going forward.
About Dystrophin
Dystrophin, a large structural protein, is critical to the stability of myofiber membranes in skeletal, diaphragmatic and cardiac muscle, protecting muscle fibers from contraction-induced damage. Loss of functional dystrophin destabilizes the dystroglycan protein complex, impairing its localization to the muscle membrane, and compromising the integrity of the membrane structure. The absence of functional dystrophin results in muscle membrane breakdown with muscle fibers being replaced by adipose and fibrotic tissue.
Eteplirsen
Eteplirsen uses phosphorodiamidate morpholino oligomer (PMO)-based chemistry and proprietary exon-skipping technology to skip exon 51 of the dystrophin gene enabling the repair of specific genetic mutations that affect approximately 13 percent of the total DMD population. By skipping exon 51, eteplirsen may restore the gene's ability to make a shorter, but still functional, form of dystrophin from messenger RNA, or mRNA. Promoting the synthesis of a truncated dystrophin protein is intended to improve, stabilize or significantly slow the disease process and prolong and improve the quality of life for patients with DMD.
About the 6-Minute Walk Test
The 6-minute walk test (6MWT) was developed as an integrated assessment of cardiac, respiratory, circulatory, and muscular capacity (American Thoracic Society 2002) for use in clinical trials of various cardiac and pulmonary conditions.
In recent years the 6MWT has been adapted to evaluate functional capacity in neuromuscular diseases and has served as the basis for regulatory approval of a number of drugs for rare diseases, with mean changes in the 6MWT ranging from 28 to 44 meters (Rubin 2002, Wraith 2004, Muenzer 2006).
Additionally, published data from longitudinal natural history studies assessing dystrophinopathy, a disease continuum comprised of DMD and Becker muscular dystrophy, support the utility of the 6MWT as a clinically meaningful endpoint (McDonald C, et al, Muscle & Nerve, December 2010) in DMD.
These data show that boys with DMD experience a significant decline in walking ability compared to healthy boys over one year, suggesting that slowing the loss of walking ability is a major treatment goal.
About the Statistical Methodology
The Mixed Model Repeated Measures (MMRM) test was used for all statistical analyses of the 6MWT results, including for all subgroups. Analysis of Covariance (ANCOVA) for ranked data was used when the assumptions of normality of the dependent variable (the change in 6MWT distance from baseline) were violated.
The inclusion of the two patients with extreme scores due to rapid progression in the ITT population (n=12) resulted in a violation of the normality assumptions of the Change from Baseline in 6MWT data, and thus required the use of ANCOVA for ranked data.
The exclusion of these two patients from the mITT population (n=10) resulted in the 6MWT data becoming normally distributed and the MMRM statistics exhibiting much improved residuals and fit statistics as compared to the ITT population.
As such, the estimated mean values and their associated p-values for the mITT population were slightly different from those for the ITT population.
Sources:
SAREPTA
Action Duchenne
Other resources:
SAREPTA will hold a conference call and broadcast a slide show today at 8:00 a.m. EDT (5:00 a.m. PDT) to discuss these results. The audio conference call may be accessed by dialing 866.356.3093 for domestic callers and 617.597.5381 for international callers. The passcode for the call is 93880948. Please specify to the operator that you would like to join the "Sarepta Therapeutics 48-Week Results Call." To view the slide show while using the audio dial-in please go to the events section of Sarepta's website at www.sareptatherapeutics.com. The call and slide show will also be webcast live under the events section and will be archived there following the call for 90 days. Please connect to Sarepta's website several minutes prior to the start of the broadcast to ensure adequate time for any software download that may be necessary. An audio replay will be available through October 10, 2012 by calling 888.286.8010 or 617.801.6888 and entering access code 67898748.
Read More
- Details
- ICNA
- News
- Hits: 807
 In an article published in the Lancet on September 27, Professor Newton and Professor Hector Garcia, both Wellcome Trust Senior Research Fellows describes the burden of epilepsy in in poor areas of the world.
In an article published in the Lancet on September 27, Professor Newton and Professor Hector Garcia, both Wellcome Trust Senior Research Fellows describes the burden of epilepsy in in poor areas of the world.
They conducted a comprehensive review of academic articles about epilepsy in developing countries in order to piece together a picture of the burden of the disease in poorer parts of the world.
The burden of epilepsy in low-income countries is more than twice that found in high-income countries, probably because the incidence of risk factors for eg head trauma, complications during childbirth, and parasite infections such as pork tapeworm (neurocysticercosis), and river blindness (onchocerciasis) is higher.
The researchers call for greater recognition from international and national health agencies to address the management of epilepsy in the developing world.
Epilepsy is associated with substantial stigma in low-income countries, which acts as a barrier to patients accessing biomedical treatment and becoming integrated within society. Seizures can be controlled by inexpensive antiepileptic drugs, but the supply and quality of these drugs can be erratic in poor areas. Prof. Charles Newton
Prof. Charles Newton
The treatment gap for epilepsy is high (>60%) in deprived areas, but this could be reduced with low-cost interventions. Despite being one of the most cost-effective disorders to treat, there are twice as many people living with epilepsy in low- and lower-middle-income countries than higher income nations and more than 60% of those affected in these regions are not accessing any appropriate treatment.
Epilepsy needs to be brought into the agenda of non-communicable diseases. It was not mentioned in the UN General Assembly Meeting in New York to address the global burden of non-communicable diseases, and yet it represents a substantial burden of ill health- Professor Charles Newton
"Sadly, adequate facilities for diagnosis, treatment and on-going management of epilepsy are virtually non-existent in many of the world's poorest regions. Many people with epilepsy or their families do not even know that they have a disorder that can be controlled with biomedical treatment, so it is vitally important that awareness is raised and medical care improved in these regions," added Professor Newton. who works in the Wellcome Trust programmes in Tanzania and Kenya
The authors call for greater recognition from international and national health agencies to address the management of epilepsy in the developing world.
Journal article:
Charles R Newton, Hector H Garcia, Epilepsy in poor regions of the world, The Lancet, Volume 380, Issue 9848, 29 September–5 October 2012, Pages 1193-1201, ISSN 0140-6736, 10.1016/S0140-6736(12)61381-6. (http://www.sciencedirect.com/science/article/pii/S0140673612613816)
Read More
- Details
- ICNA
- News
- Hits: 759
 Children suffering from prolonged, acute, convulsive seizures may not always receive timely rescue medication in schools and other community settings as intended by their specialist physician, according to the first findings of the PERFECT[1] Initiative. The results were presented as part of a symposium at the ILAE's 10th European Congress on Epileptology (ECE), in London. The Steering Committee for The PERFECTTM Initiative, which comprises a group of leading clinical epilepsy specialists from six countries across Europe, also highlight discrepancies in comprehensive European guidelines and legal frameworks that ensure children with prolonged, acute, convulsive seizures are treated quickly whether in hospital or in the community, and recommend specific training on rescue medication for all those responsible for the child.[i]
Children suffering from prolonged, acute, convulsive seizures may not always receive timely rescue medication in schools and other community settings as intended by their specialist physician, according to the first findings of the PERFECT[1] Initiative. The results were presented as part of a symposium at the ILAE's 10th European Congress on Epileptology (ECE), in London. The Steering Committee for The PERFECTTM Initiative, which comprises a group of leading clinical epilepsy specialists from six countries across Europe, also highlight discrepancies in comprehensive European guidelines and legal frameworks that ensure children with prolonged, acute, convulsive seizures are treated quickly whether in hospital or in the community, and recommend specific training on rescue medication for all those responsible for the child.[i]
Prolonged, acute, convulsive seizures can pose a significant health threat in children with epilepsy, a neurological disorder affecting nearly one million children and adolescents in Europe.[ii] Evidence suggests that treatment should be given immediately if a seizure persists longer than 5 minutes after onset.[iii] However, in the case of schools, despite the fact that many children are prescribed rescue medication by their doctors, teachers often opt not to administer seizure rescue medication unless specific training or provision has been made, typically via the school nurse. Instead an ambulance may be called, causing possible delays in seizure treatment.[iv]
"The PERFECT Initiative is the first to investigate the discrepancies that often exist in European countries between policy and practice in the treatment of prolonged, acute, convulsive seizures in children," said Prof J. Helen Cross, UCL Institute of Child Health, Great Ormond Street Hospital for Children and Young Epilepsy. "We found that the differences in clear guidance, awareness and education around the use of rescue medication for treating seizures in children living with epilepsy, ultimately create a shortfall in care that we, as clinicians, intend that they receive, whether in hospital or away from it."
This first phase of the PERFECT Initiative was designed to examine existing treatment guidelines and legal frameworks and policies for treating prolonged, acute, convulsive seizures in the community, in six European countries (France, Germany, Italy, Spain, Sweden and the UK). The authors found that, while guidelines were effective for in-hospital treatment of prolonged, acute, convulsive seizures, the picture was often different in the community. In many cases, rescue medication consists of intra-rectal diazepam, which can be considered socially inappropriate to administer in the community setting. Whether a child receives rescue medication at school depends primarily on the availability of staff willing to accept responsibility for administering the treatment.1 Key recommendations from the PERFECT Initiative Steering Committee include:1
Establishing clear links between the treating physician, families and the child's day-to-day community environment (e.g. schools), allowing for better provision of information on epilepsy and training on seizure intervention for all those individuals responsible for the child
New and revised comprehensive guidelines to ensure children with prolonged, acute, convulsive seizures are treated according to the treatment plan set by their physician, wherever the seizure occurs
Individualised treatment plans for every child are established between the treating physician and the family/carers concerned, to help ensure the best possible standards of care for the child away from the hospital setting.
ViroPharma Incorporated (Nasdaq: VPHM), as part of its commitment to help create better care for children with prolonged, acute, convulsive seizures, has organised and funded the PERFECT Initiative in collaboration with a group of leading clinical epilepsy specialists. Vanessa Newman, Associate Director Medical Affairs, Europe, at ViroPharma said: "The PERFECT Initiative was designed to investigate any possible shortfalls in the current approach in Europe to the management of prolonged, acute, convulsive, seizures in children treated in the community with rescue medication. We believe that we can help improve care for children at risk of seizures by supporting collaboration between key stakeholders, identifying opportunities for social integration, and facilitating better education at all levels. We are proud to have initiated PERFECT and look forward to helping support the improvement in care for children with epilepsy that The Steering Committee recommends."
The second two phases of the PERFECT Initiative include a survey of physicians and nurses who treat children with prolonged, acute, convulsive seizures, followed by a survey of children and their parents regarding their treatment, care, guidance and quality of life. Results of these phases are due to be published in 2013.
About the PERFECT Initiative
ViroPharma, as part of its commitment to help create better care for children with prolonged, acute, convulsive seizures, has organised and funded the PERFECT (Practices in Emergency and Rescue medication for Epilepsy managed with Community administered Therapy) Initiative in collaboration with a Steering Committee group of leading clinical epilepsy specialists. The Initiative aims to document and communicate first of its kind data on the impact of conflict of policy and practice in the treatment of prolonged, acute, convulsive seizures.
The PERFECT Initiative is designed to facilitate collaboration between key stakeholders, identifying opportunities for social integration around children experiencing breakthrough seizures, and supporting means of better education of the condition at the community level. It consists of three phases, a review and comparison of policy and real-world practice, a survey of physicians and nurses who treat children with prolonged, acute, convulsive seizures, and a survey of children and their carers relating to their experiences of breakthrough seizures.
References:
[1] Practices in Emergency and Rescue medication For Epilepsy managed with Community administered Therapy
[i] Wait S, et al. The administration of rescue medication to children with prolonged acute convulsive seizures in the community: what happens in practice? Eur J Paediatr Neurol. 2012. doi:10.1016/j.ejpn.2012.07.002. Published early online.
[ii] Epilepsy in the WHO European Region: Fostering Epilepsy Care in Europe. 2010. Available at: http://www.ibe-epilepsy.org/downloads/EURO%20Report%20160510.pdf. Last accessed July 2012.
[iii] Lagae L. The treatment of acute convulsive seizures in children. Eur J Pediatr 2011;170:413-8. [iv] Kriel RL, et al. Home use of rectal diazepam for cluster and acute prolonged seizures: efficacy, adverse reactions, quality of life, and cost analysis. Pediatr Neurol 1991;7:13-17.
[v] Ekinci O, et al. Depression and anxiety in children and adolescents with epilepsy: Prevalence, risk factors, and treatment. Epilepsy Behav 2009;14:8-18.
Read More