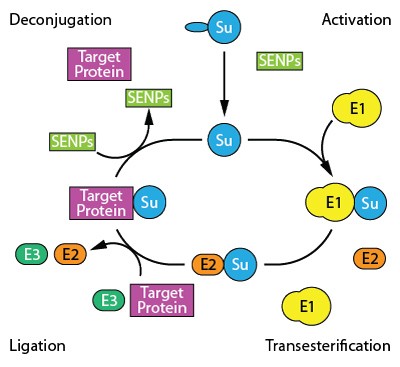
In a study published in The FASEB Journal a team of NIH-funded scientists reports on a potential drug that could be protective in ischemic stroke patients.
The clue for the study came from observations in hibernating ground squirrels. In these animals there is a dramatic reduction of cerebral blood flow during hibernation, yet they emerge from their sleep without any ill effects at all.
The researchers observed that a cellular process called SUMOylation goes into overdrive in a certain species of ground squirrel during hibernation. They suspected this might be the mechanism by which the animals' brains survived the reduced blood flow caused by hibernation, and subsequent experiments in cells and mice confirmed this suspicion.Reference:
Bernstock et al. Quantitative high-throughput screening identifies cytoprotective molecules that enhance SUMO-conjugation via the inhibition of SUMO-specific protease (SENP)2. The FASEB Journal. November 16, 2017. doi: 10.1096/fj.201700711R.

SUMOylation: A Post-translational Modification Targeting Cytoskeletal Proteins
Read More
- Details
- ICNA
- News
- Hits: 1100

New research published in Epilepsia, a journal of the International League Against Epilepsy (ILAE), indicates that wristband devices may improve the detection and characterization of seizures in patients with epilepsy.
In their attempts to develop a better monitoring method, Giulia Regalia, PhD and Francesco Onorati, PhD, of Empatica Inc. in Milan, Italy and Cambridge, Massachusetts, and their colleagues examined the potential of automated, wearable systems to detect and characterize convulsive epileptic seizures. The researchers used three different wristbands to record two signals -- called electrodermal activity and accelerometer signals -- that usually exhibit marked changes upon the onset of convulsive seizures, obtaining 5928 hours of data from 69 patients, including 55 convulsive epileptic seizures from 22 patients.
The wristband detectors showed high sensitivity (95% of seizures were detected) while keeping the false alarm rate at a bearable level (on average, one false alarm every four days), which improves a pioneering 2012 study led by MIT professor Rosalind Picard, now chief scientist at Empatica.
In addition to detecting seizures, the method also revealed certain characteristics of the seizures, which may help alert clinicians and patients to seizures that are potentially dangerous and life-threatening.
"Multi-center clinical assessment of improved wearable multimodal convulsive seizure detectors." Francesco Onorati, Giulia Regalia, Chiara Caborni, Matteo Migliorini, Daniel Bender, Ming-Zher Poh, Cherise Frazier, Eliana Kovitch Thropp, Elizabeth D. Mynatt, Jonathan Bidwell, Roberto Mai, W. Curt LaFrance, Jr., Andrew S. Blum, Daniel Friedman, Tobias Loddenkemper, Fatemeh Mohammadpour-Touserkani, Claus Reinsberger, Simone Tognetti, and Rosalind W. Picard. Epilepsia; Published Online: October 4, 2017 (DOI: 10.1111/epi.13899).
Read More
- Details
- ICNA
- News
- Hits: 1362
This largest whole-genome sequencing study on epilepsy was piloted by Jacques Michaud, Pediatrician at CHU Sainte-Justine and Professor of Pediatrics and Neuroscience at the Faculty of Medicine of Université de Montreal and his colleagues, Elsa Rossignol and Patrick Cossette of Universite de Montréal and Berge Minassian of the University of Toronto.
The team identified eight new genes involved in this type of epilepsy thanks to their use of whole-genome sequencing, which had never been done before in an epileptic study of this scope. The results of their study were recently published in the American Journal of Human Genetics. The researchers have identified a causal link between Developmental and Epileptic Encephalopathy (DEE) and the following genes: NTRK2, GABRB2, CLTC, DHDDS, NUS1, RAB11A, GABBR2, and SNAP25. Developmental and epileptic encephalopathy (DEE) is a group of conditions characterised by the co-occurrence of epilepsy and intellectual disability (ID), typically with developmental plateauing or regression associated with frequent epileptiform activity.
This discovery has further-reaching implications. In the context of epilepsy de novo mutations seem to involve mechanisms of gene disruption that are unlike those involved in intellectual disability. Mutations in epilepsy tend to affect specific areas of the gene, whereas mutations associated with intellectual disability are more often distributed throughout the entire gene.Knowledge of these mechanisms of action is crucial for the development of personalised epilepsy treatments. However, much more work is needed before these treatments can be harmonised with patients' genetic profiles.
Source: AAAS
High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies
Hamdan, Fadi F. et al.The American Journal of Human Genetics , Volume 101 , Issue 5 , 664 - 685
http://dx.doi.org/10.1016/j.ajhg.2017.09.008
Read More
- Details
- ICNA
- News
- Hits: 971
In a study published in Neonatology on Oct. 13, 2017, researchers from George Washington University, Washington, DC using newborn piglet models exposed to hypoxia-ischemia studied the ffect of standard cooling therapy (therapeutic hypothermia) alone and in combination with a selective Src kinase inhibitor, PP2, that blocks a regulatory enzyme of apoptosis (cell death). PP2 is a substance that has frequently been used in cancer research as a "selective" inhibitor for Src-family kinases.
This study is the first to test the benefits of blocking this enzyme in reducing the neurological damage caused by brain hypoxia-ischemia. A Src kinase inhibitor is already approved by FDA as an oncology treatmentIn hypoxia-ischemia, CaM kinase is over-activated, but hypothermia has been shown to decrease this enzyme's activation. They hypothesized that a Src kinase inhibitor, in addition to hypothermia, would further attenuate the activation of CaM kinase IV and that the result might be less brain damage.
The research team assessed neuropathology, adenosine triphosphate and phosphocreatine concentrations as well as CaM kinase IV activity. The CaM kinase IV activity in cerebral tissue was 2,002 (+/- 729) with normal oxygen levels and in normal temperatures, 4,104 (+/- 542) in hypoxia with hypothermia treatment, and 2,165 (+/- 415) in hypoxia with hypothermia treatment combined with PP2 administration.
The authors conclude that hypothermia alone attenuated the over-activation of CaM kinase IV and improved neuropathology after hypoxia. However, the combination of hypothermia with Src kinase inhibition following hypoxia further attenuated the increased activation of CaM kinase IV, compared with hypothermia alone in the newborn swine brain.
At present therapeutic hypothermia remains the mainstay of treatment for hypoxia-ischemia is .This therapy is proven to reduce neural defects by up to 30 percent, yet many infants still have poor outcomes even after the therapeutic cooling treatment.
If confirmed by further studies, this approach in combination with cooling may help to further attenuate neurological damage following hypoxic ischemic injuries.
The group intends to study the effect of other types of small molecule inhibitors to target the apoptotic cascade.
Effect of Concurrent Src Kinase Inhibition with Short-Duration Hypothermia on Ca2+/Calmodulin Kinase IV Activity and Neuropathology after Hypoxia-Ischemia in the Newborn Swine Brain
Kratimenos P et al. Neonatology 2018;113:37-43
https://doi.org/10.1159/000480067
Read More
- Details
- ICNA
- News
- Hits: 942

In an article published in the Journal of Neuroscience Prof. John Chatham, of the Department of Pathology at the University of Alabama at Birmingham, and colleagues report that increasing O-GlcNAcylation levels in brain cells using the dietary supplement "glucosamine" widely used as a supplement to help reduce pain in osteoarthritis and other conditions was found to reduce reduce neural excitability in rodents.
The researchers had in a previous study shown that increases in protein O-GlcNAcylation are associated with a reduction in the strength of synapses in the hippocampus of the brain. This synaptic dampening effect on glutamatergic networks suggests that increasing O-GlcNAcylation will depress pathological hyperexcitability. Using in vitroand in vivo models of epileptiform activity, they have shown in the current study that acutely increasing O-GlcNAc levels can significantly attenuate ongoing epileptiform activity and prophylactically dampen subsequent seizure activity.
They tested this hypothesis using glucosamine which blocks an enzyme that clears O-GlcNAcylation from the brain, leading to a rapid increase in levels of the protein. Glucosamine was applied to hippocampal brain slices derived from rats and mice in which epileptiform activity was generated by GABAAR inhibition using drugs. The drug induced neural excitability was found to be dampened with ten minutes of the application of the compound glucosamine.
This is an exciting finding representing another target for research in epilepsy therapeutics.
Stewart LT, Khan AU, Wang K, Pizarro D, Pati S, Buckingham SC et al. (2017) Acute Increases in Protein O-GlcNAcylation Dampen Epileptiform Activity in Hippocampus.J Neurosci 37 (34):8207-8215. DOI: 10.1523/JNEUROSCI.0173-16.2017 PMID: 28760863.
Read More
- Details
- ICNA
- News
- Hits: 1549