 The Myotubular Trust announces A Fifth Call To Grant Application. The trust is looking to fund further projects that will help find a cure and/or a treatment for any of the three types of myotubular myopathy (congenital X-linked recessive; congenital autosomal recessive; autosomal dominant), focusing on research that would not generally be funded by public or industrial funding sources. The call is open to research bodies internationally.
The Myotubular Trust announces A Fifth Call To Grant Application. The trust is looking to fund further projects that will help find a cure and/or a treatment for any of the three types of myotubular myopathy (congenital X-linked recessive; congenital autosomal recessive; autosomal dominant), focusing on research that would not generally be funded by public or industrial funding sources. The call is open to research bodies internationally.
To date, the trust have awarded six research grants / fellowships for the following projects:
- Gene therapy for x-linked myotubular myopathy and pathophysiology – Dr Anna Buj Bello, Genethon, Evry
- Membrane trafficking and T tubule structure and function in a canine model of centronuclear myopathies – Dr Richard Piercy, Royal Veterinary College, London
- Deciphering the molecular pathway involving centronuclear myopathy genes – Manuela D’Alessandro, IGBMC, Illkirch
- Gene therapy for autosomal dominant centronuclear myopathy by Transplicing – Dr Marc Bitoun, INSERM, Paris
- Next generation sequencing to tackle centronuclear myopathies – Dr Jocelyn Laporte, IGBMC, France
- Secondary pathogenic mechanisms in XLMTM and CNM – Dr Susan Treves, Basel University Hospital, Basel and Dr Heinz Jungbluth, King’s College London
In particular the trust encourages the application of new technologies to research into myotubular myopathy, which may involve collaboration between different medical disciplines and / or different research institutions. They are also willing to consider applications which involve joint funding with other organisations.
Please see the Myotubular trust Research Programme & Grants Information Page to read more and download an application form.
Source: The Myotubular Trust
The Myotubular Trust is not affiliated to The International Child Neurology Association (ICNA)
Read More
- Details
- ICNA
- News
- Hits: 683
 A European team of scientists have built the first atlas of white-matter microstructure in the human brain. The project’s final results have the potential to change the face of neuroscience and medicine over the coming decade.
A European team of scientists have built the first atlas of white-matter microstructure in the human brain. The project’s final results have the potential to change the face of neuroscience and medicine over the coming decade.
The work relied on groundbreaking MRI technology and was funded by the EU’s future and emerging technologies program with a grant of 2.4 million Euros. The participants of the project, called CONNECT, were drawn from leading research centers in countries across Europe including Israel, United Kingdom, Germany, France, Denmark, Switzerland and Italy.
The project investigators met on October 19, 2012 in Paris, after 3 years of research, to announce the conclusion of the project and present a report of their findings.
The new atlas combines three-dimensional images from the MRI scans of 100 brains of volunteers. To achieve this, CONNECT developed advanced MRI methods providing unprecedented detail and accuracy.
 Professor Daniel Alexander, a CONNECT steering committee member from the UCL Department of Computer Science said: "The UCL team use the latest computer modelling algorithms and hardware to invent new imaging techniques. The techniques we devised were key to realising the new CONNECT brain atlas."The imaging techniques reveal new information about brain structure that help us understand how low-level cellular architecture relate to high-level thought processes."
Professor Daniel Alexander, a CONNECT steering committee member from the UCL Department of Computer Science said: "The UCL team use the latest computer modelling algorithms and hardware to invent new imaging techniques. The techniques we devised were key to realising the new CONNECT brain atlas."The imaging techniques reveal new information about brain structure that help us understand how low-level cellular architecture relate to high-level thought processes."
Currently, biomedical research teams around the world studying brain science rely on a brain atlas produced by painstaking and destructive histological methods on the brains of a few individuals who donated their bodies to science.
The new atlas simulates the impossible process of painstakingly examining every mm2 of brain tissue (of which there are around 100 million per brain) with a microscope, while leaving the brain in tact.
The key novelty in the atlas is the mapping of microscopic features (such as average cell size and packing density) within the white matter, which contains the neuronal fibers that transmit information around the living brain. The results of the project, obtained through advanced image processing techniques, provide new depth and accuracy in our understanding of the human brain in health and disease.
The atlas describes the brain's microstructure in standardized space, which enables non-expert users, such as physicians or medical researchers, to exploit the wealth of knowledge it contains. The atlas contains a variety of new images that represent different microscopic tissue characteristics, such as the fiber diameter and fiber density across the brain, all estimated using MRI. These images will serve as the reference standard of future brain studies in both medicine and basic neuroscience.
The project will dramatically facilitate and promote future research into white matter structure and function. Historically in neuroscience, the vast majority of research effort has been invested in understanding and studying gray matter and neurons, while white matter has received relatively little attention.
This owes largely to the lack of effective research tools to study white matter, even though it comprises about half the volume of the brain. The new MRI methods that were developed in CONNECT allow researchers, for the first time, to visualize the micro-structure of the living brain over the whole brain.
This opens new realms in our understanding of our most complex organ. In the future, the project members intend to use the technology they have developed to study the dynamics and time dependence of the micro-structure in white matter. For example they will search for a finger print or a trace that a cognitive task imprints on white matter microstructure encoding new experiences in the wiring of the brain.
Another future direction is to characterize and understand micro-structural changes caused by different neurodegenerative diseases, such as Alzheimer's or schizophrenia, in order to develop better diagnostic procedures for these and other devastating conditions.
Source: University College London
Read the full CONNECT Report here or view below
Read More
- Details
- ICNA
- News
- Hits: 745
 The 18th Mediterranean and Pan Arab Child Neurology Congress In Collaboration with the Egyptian Society of Child Neuropsychiatry (ESCNP) will be held at Hilton Green Plaza Alexandria, Egypt from April 17-18, 2013. The deadline for submission of abstracts is February 15, 2013. Abstracts are to be send as a word document (A4, Arial Size 12) to This email address is being protected from spambots. You need JavaScript enabled to view it.
The 18th Mediterranean and Pan Arab Child Neurology Congress In Collaboration with the Egyptian Society of Child Neuropsychiatry (ESCNP) will be held at Hilton Green Plaza Alexandria, Egypt from April 17-18, 2013. The deadline for submission of abstracts is February 15, 2013. Abstracts are to be send as a word document (A4, Arial Size 12) to This email address is being protected from spambots. You need JavaScript enabled to view it.
| Before March 15, 2013 | After March 15, 2013 & Onsite | |
| Congress Registration | 150 USD | 200 USD |
| Workshop | 50 USD | 80 USD |
| Packages | ||
| SINGLE | 2000 LE | 2400 LE |
| DOUBLE | 2800 LE | 3200 LE |
|
Packages include |
||
Pan Arab Committee
Mohamed Jan, KSA
Suad Al Yamani, KSA
Hicham Mansour, Lebanon
Samir Bakleh, Syria
Omar Ismail, Syria
Abdel Rahamn Ishak, Yemen
Abdel Karim Al Qudah, Jordan
Mostafa Saleh, Sudan
Haydar Babikir, Sudan
Nagwa Milady, Tunisia
Shahnaz Triki, Tunisia
Noureddine Djebl, Algeria
ESCNP Committee
Ahmed Raouf President
Ahmed Younis Vice President
Nabil Kitchener General Secretary
Nagwa Abdel Meguid Treasurer
Members
Emad Hammad
Hussein Hosny Abdeldayem
Ola Shahin
Sherif Abd Elaal
Shora Yousef
Organized by
Cairo Med Egypt CME
http://www.cme-group.net
Read More
- Details
- ICNA
- News
- Hits: 885
 A study led by Brian McCabe and his team from the Motor Neuron Center at Columbia University Medical Center (CUMC) suggests that contrary to existing theories, Spinal muscular atrophy (SMA), results primarily from motor circuit dysfunction and not motor neuron or muscle cell dysfunction
A study led by Brian McCabe and his team from the Motor Neuron Center at Columbia University Medical Center (CUMC) suggests that contrary to existing theories, Spinal muscular atrophy (SMA), results primarily from motor circuit dysfunction and not motor neuron or muscle cell dysfunction
SMA, a hereditary neuromuscular disease characterized by muscle atrophy and weakness is caused by defects in a gene called SMN1 (survival motor neuron 1), which encodes the SMN protein.
There are several forms of SMA, distinguished by time of onset and clinical severity. The most severe form, Type 1, appears before six months of age and generally results in death by age two. In milder forms, symptoms may not appear until much later in childhood or even in early adulthood. There is no treatment for SMA, which is estimated to affect as many as 10,000 to 25,000 children and adults in the United States and is the leading genetic cause of death in infants.
The researchers studied1 Drosophila (Fruit Flies) SMN mutants which had been genetically altered so that every cell had a defective copy of the SMN1 gene. The mutant flies had reduced muscle size and defective locomotion, motor rhythm, and motor neuron neurotransmission. When fully functional copies of SMN1 were introduced into the flies' motor neurons or muscle cells, the cell types previously thought to be affected, the flies unexpectedly did not show any improvement.
 However when SMN1 was returned to other motor circuit neurons - in particular, proprioceptive neurons and interneurons – the muscle size and motor function were restored. The proprioceptive neurons in motor circuits pick up and relay information to the spinal cord and brain about the body's position in space, which is then processed in the CNS and relayed via interneurons back to the motor neurons to stimulate muscle movement.
However when SMN1 was returned to other motor circuit neurons - in particular, proprioceptive neurons and interneurons – the muscle size and motor function were restored. The proprioceptive neurons in motor circuits pick up and relay information to the spinal cord and brain about the body's position in space, which is then processed in the CNS and relayed via interneurons back to the motor neurons to stimulate muscle movement.
In another experiment the researchers demonstrated that in fruit flies with defective SMN1, proprioceptive neurons and interneurons do not produce enough neurotransmitters. The muscle size and motor function improved when the flies' potassium channels were genetically blocked thereby increasing neurotransmitter output. The same effect was seen when the flies were given drugs that block potassium channels, leading to suggestions that this class of drugs might help patients with SMA.
Supported by these findings, in July 2012, the SMA Clinical Research Center at CUMC launched a clinical trial of a potassium channel blocker called dalfampridine (Ampyra) for the treatment of patients with SMA.
The study will assess whether the Dalfampriine improves walking ability and endurance in adults with SMA Type 3, compared with placebo. Claudia A. Chiriboga, MD, MPH, associate professor of Clinical Neurology at CUMC, is the lead clinical investigator. Ampyra was approved by the FDA for the treatment of patients with multiple sclerosis in 2010.
In a second study2, led jointly by Livio Pellizzoni, PhD, assistant professor of Pathology and Cell Biology in the Motor Neuron Center, and Dr. McCabe looked at why even though mutations in the disease gene SMN1 results in reduced expression in all cells, its only the motor system that is affected in patients with SMA.
Working with models of SMA in mammalian cells, fruit flies, zebrafish, and mice, the researchers demonstrated that SMN1 deficiency disrupts a fundamental cellular process known as RNA splicing (removal of parts of RNA called introns so that a gene can be translated into protein) with detrimental effects on the expression of a subset of genes that contain a rare type of intron.
 By studying the function of this group of genes affected by the loss of SMN1, the researchers discovered a novel gene - which they named stasimon - that is critically required for motor circuit activity in vivo. They further showed that restoring expression of stasimon was alone sufficient to correct key aspects of motor dysfunction in both invertebrate and vertebrate models of SMA.
By studying the function of this group of genes affected by the loss of SMN1, the researchers discovered a novel gene - which they named stasimon - that is critically required for motor circuit activity in vivo. They further showed that restoring expression of stasimon was alone sufficient to correct key aspects of motor dysfunction in both invertebrate and vertebrate models of SMA.
The implication is that this gene and the pathway in which it functions might be new candidate therapeutic targets The loss of the SMN1 gene has been directly linked to defective splicing of a critical neuronal gene to motor circuit dysfunction. The study thus points to SMA being a disease of RNA splicing.
References:
Read More
- Details
- ICNA
- News
- Hits: 708
 A Phase I clinical trial led by investigators from the University of California, San Francisco (UCSF) has shown that neural stem cells successfully engrafted into the brains of patients and appear to have produced myelin. The findings have been published in the Oct 10, 2012 issue of Science Translational Medicine.
A Phase I clinical trial led by investigators from the University of California, San Francisco (UCSF) has shown that neural stem cells successfully engrafted into the brains of patients and appear to have produced myelin. The findings have been published in the Oct 10, 2012 issue of Science Translational Medicine.
Once transplanted and engrafted, neural stem cells have the potential to differentiate into a number of different brain cell types, depending on the area of the brain into which they are inserted. The sites chosen for the Phase I study were known from animal studies to be the most likely to result in the formation of oligodendrocytes.
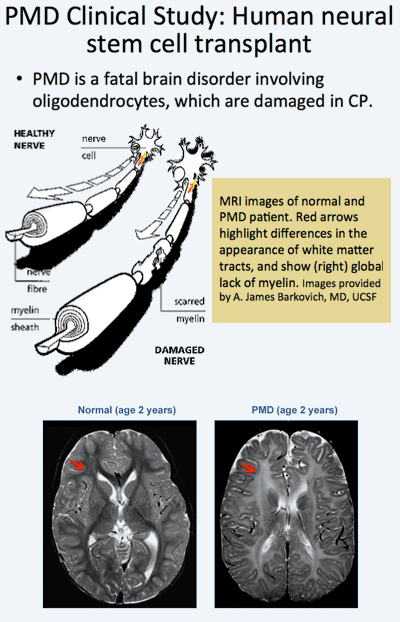 In the trial, allogeneic human neural stem cells (HuCNS-SCs) developed by Stem Cells, Inc., of Newark, California, were injected directly into the frontal lobe white matter of four young children with an early-onset, fatal form of a condition known as Pelizaeus-Merzbacher disease (PMD).
In the trial, allogeneic human neural stem cells (HuCNS-SCs) developed by Stem Cells, Inc., of Newark, California, were injected directly into the frontal lobe white matter of four young children with an early-onset, fatal form of a condition known as Pelizaeus-Merzbacher disease (PMD).
Immunosuppression was administered for 9 months. During 2010-2011, the children with PMD, who were included in the trial, underwent serial neurological evaluations, developmental assessments, and cranial magnetic resonance imaging (MRI) and MR spectroscopy, including high-angular resolution diffusion tensor imaging (DTI). The investigators found evidence that the stem cells had successfully engrafted, receiving blood and nutrients from the surrounding tissue and integrating into the brain.
The Phase 1 trials designed to test safety and preliminary efficacy demonstrated that the neural stem cells were safe in the patients’ brains one year post transplant. No clinical or radiological adverse effects were directly attributed to the donor cells. Reduced T1 and T2 relaxation times were observed in the regions of transplantation 9 months after the procedure in the three subjects. Normalized DTI showed increasing fractional anisotropy and reduced radial diffusivity, consistent with myelination, in the region of transplantation compared to control white matter regions remote to the transplant sites. The MRI findings suggested myelination in the regions that have been transplanted providing indirect evidence that the stem cells had become oligodendrocytes and were producing myelin.
 These results have thus provided for the first time evidence that transplanted neural stem cells are able to produce new myelin in patients with a severe myelination disease. The MRI findings from the clinical trial are further supported by findings from a separate study2 by in a separate study by researchers at Oregon Health & Science University's Papé Family Pediatric Research Institute which showed that neural stem cells injected into mouse models became oligodendrocytes and formed myelin.
These results have thus provided for the first time evidence that transplanted neural stem cells are able to produce new myelin in patients with a severe myelination disease. The MRI findings from the clinical trial are further supported by findings from a separate study2 by in a separate study by researchers at Oregon Health & Science University's Papé Family Pediatric Research Institute which showed that neural stem cells injected into mouse models became oligodendrocytes and formed myelin.
 The PMD clinical trial researchers included Principal investigator David H. Rowitch, MD, PhD, a professor of pediatrics and neurological surgery at UCSF, chief of neonatology at UCSF Benioff Children's Hospital and a Howard Hughes Medical Institute Investigator and co-principal investigator Nalin Gupta, MD, PhD, associate professor of neurological surgery and pediatrics and chief of pediatric neurological surgery at UCSF Benioff Children's Hospital.
The PMD clinical trial researchers included Principal investigator David H. Rowitch, MD, PhD, a professor of pediatrics and neurological surgery at UCSF, chief of neonatology at UCSF Benioff Children's Hospital and a Howard Hughes Medical Institute Investigator and co-principal investigator Nalin Gupta, MD, PhD, associate professor of neurological surgery and pediatrics and chief of pediatric neurological surgery at UCSF Benioff Children's Hospital.
The study, one of the first neural stem cell trials ever conducted in the United States, is symbolical of UCSF’s pioneering role in the stem cell field.
![]() In 1981, Gail Martin, PhD, professor of anatomy, co-discovered embryonic stem cells in mice.
In 1981, Gail Martin, PhD, professor of anatomy, co-discovered embryonic stem cells in mice.
![]() In 2001, Roger Pedersen, PhD, professor emeritus of obstetrics, gynaecology and reproductive sciences, derived two of the first human embryonic stem cell lines.
In 2001, Roger Pedersen, PhD, professor emeritus of obstetrics, gynaecology and reproductive sciences, derived two of the first human embryonic stem cell lines.
![]() In 2012, Shinya Yamanaka, MD, PhD, of the UCSF-affiliated Gladstone Institutes and Kyoto University, received the Nobel Prize in Physiology or Medicine for his discovery that adult cells can be reprogrammed to behave like embryonic stem cells
In 2012, Shinya Yamanaka, MD, PhD, of the UCSF-affiliated Gladstone Institutes and Kyoto University, received the Nobel Prize in Physiology or Medicine for his discovery that adult cells can be reprogrammed to behave like embryonic stem cells
Citations
1Gupta N, Henry RG, Strober J, Kang SM, Lim DA, Bucci M et al. (2012) Neural stem cell engraftment and myelination in the human brain. Sci Transl Med 4 (155):155ra137. DOI: 10.1126/scitranslmed.3004373 PMID: 23052294.
2Uchida N, Chen K, Dohse M, Hansen KD, Dean J, Buser JR et al. (2012) Human neural stem cells induce functional myelination in mice with severe dysmyelination. Sci Transl Med 4 (155):155ra136. DOI: 10.1126/scitranslmed.3004371 PMID: 23052293.
About Pelizaeus-Merzbacher disease (PMD)
- Rare congenital X-linked recessive leukodystrophy
- Incidence of 1:200,000 to 1:500,000
- Caused by mutation of myelin protein proteolipid protein 1 (PLP1), resulting in hypomyelination
- Leading to death between ages 10 and 15
- Oligodendrocytes are unable to myelinate axons, resulting in loss of normal axonal conduction and neurological dysfunction in the short term, eventually leading to axonopathy and neurodegeneration.
- PMD is one of a spectrum of diseases associated with PLP1, which also includes Spastic Paraplegia Type 2 (SPG2)
- Four types recognised
- Congenital PMD -early-onset severe form of PMD presents with profound neurodevelopmental deficits.
- Classic PMD, in which the early symptoms include muscle weakness, involuntary movements of the eyes (nystagmus), and delays in motor development within the first year of life;
- Complicated SPG2, which features motor development issues and brain involvement, and,
- Pure SPG2, which includes cases of PMD that do not have neurologic complications.
Further Reading
Pelizaeus-Merzbacher disease (PMD)
Read More
- Details
- ICNA
- News
- Hits: 721
- ICNA Educational Meeting in Pune January 19-20, Pune, India
- Eteplirsen trial results announced- potential breakthrough in DMD treatment
- The Burden of Epilepsy in Low Income Countries reviewed
- PERFECT Initiative Shows That Children With Epilepsy May Not Be Receiving Treatment For Prolonged, Acute, Convulsive Seizures