
Background
Although there is much information on the internet about epilepsy and seizures, there is a glaring absence of a single source of information that aligns with the international classification and provides an organized presentation of the many seizure types and syndromes to help with diagnosis and treatment. This information gap was recognized and led to the ILAE's EpilepsyDiagnosis.Org project which was formally launched in September 2014. It has been a unique resource in medicine and has harnessed the power of the internet to present the complexity of the significant amount of new information now available about the epilepsies and their etiologies, in a manner that is concise, current and accessible to a global audience. It is as relevant to those in primary and secondary health care settings as it is to those in tertiary epileptology practice. It is also showing promise as an instructional and training resource for those who are new to medicine.
The project EpilepsyDiagnosis.org was conceived and developed by the ILAE's Commission on Classification and Terminology (2009-2013), and this Commission's Diagnostic Manual Taskforce (Table 1), in partnership with eResearch at the University of Melbourne, Australia. The project has been further developed by the ILAE's Commission on Classification and Terminology (2013-2017), and this Commission's EpilepsyDiagnosis.Org and Syndromes Task Force (Table 1).
Since the release of EpilepsyDiagnosis.org, its reach has steadily increased, month on month. Currently approximately 10,000 unique visitors access the site each month from around the world, viewing EpilepsyDiagnosis.Org pages more than 40,000 times per month. Users of the website span professional groups that range from those in primary care to those working in tertiary health care settings (Table 1). The ongoing growth in user engagement with EpilepsyDiagnosis.Org continues to occur 'organically', through relevance of the website content to those in clinical practices where epilepsy is diagnosed, and managed.
Goals
The goals of EpilepsyDiagnosis.org are:
- to make available, in an easy to understand form, the latest concepts relating to seizures and the epilepsies.
- to assist clinicians, particularly those in primary and secondary health care settings anywhere in the world, who look after people with epilepsy to diagnose seizure type(s), classify epilepsy, diagnose epilepsy syndromes and define the etiology.
- to provide an educational resource that is current for personal learning and small group teaching settings.

What you will find on the EpilepsyDiagnosis.Org website
The structure of the site reflects the importance of seizure type, syndrome, and etiology in clinical practice, and how these aspects of the epilepsy inter-relate. On the site you will find:
- seizure type classification with video examples of seizure types – the availability of video is a unique feature of this site, allowing clinicians to clearly see the features of seizures, including distinguishing features from other similar seizure types. A short and instantaneous registration process is required to view the video section and this is open to anyone, anywhere in the world with an internet connection. Individuals and their families have kindly given consent for videos to be freely available in this way.
- seizure types presented with differential diagnoses, including a comprehensive section on epilepsy imitators – where you will find full descriptions of non-epileptic paroxysmal phenomena that can mimic seizures.
- focal seizure types flexibly described by their features, and by features that suggest anatomical localization (Figure 1).
- epilepsy syndromes presented in a comprehensive list, including details on their clinical presentations, EEG and imaging features (with images to illustrate these) and current understanding of syndrome etiologies
- epilepsy etiologies presented in a comprehensive but concise section that includes most notably genetic and structural etiologies, but also including content on metabolic and immune etiologies. The etiology section provides concise and clinically relevant information on phenotypes seen with more than 50 genes associated with epilepsy, as well as the phenotypes seen in chromosomal abnormalities associated with epilepsy.
In 2016 a significant upgrade has occurred to the structural etiologies content, making available the most current knowledge regarding brain abnormalities associated with epilepsy, especially newer information regarding their genetic bases. The site now includes a 'tour de force' of the following structural etiologies for epilepsy:
- malformations of cortical development: focal cortical dysplasia, tuberous sclerosis, lissencephaly, subcortical band heterotopia, grey matter heterotopia, polymicrogyria, hemimegalencephaly, schizencephaly and hypothalamic hamartoma
- vascular malformations: cerebral angioma, Sturge-Weber syndrome and arteriovenous malformation
- hippocampal sclerosis
- hypoxic-ischemic: stroke and hypoxic ischemic brain injury
- traumatic brain injury o tumors: dysembryoplastic neuroepithelial tumors and ganglioglioma, and
- porencephalic cysts
EpilepsyDiagnosis.Org complements resources available through Epileptic Disorders, the ILAE's official educational journal, for professionals with particular interest in epilepsy. However, EpilepsyDiagnosis.Org through its open access format, also provides an increased reach to health professionals from primary and secondary health care settings who see patients with epilepsy, and is relevant for community organizations and for the general public due to the simple and clear presentation of information.
Please visit and use this site at https://www.epilepsydiagnosis.org/. Your comments and suggestions are welcome in the Give Feedback section.
| Table 1: Visitors to EpilepsyDiagnosis.Org by professional background (top 10, accounting for 52% of all visitors) |
| Secondary Health Care - Adult Neurology 8% Secondary Health Care - Pediatrics General 7% Postgraduate Medical Trainee - Adult Medicine 6% Secondary Health Care - Pediatric Neurology 6% Tertiary Health Care - Pediatric Neurology 6% Tertiary Health Care - Adult Neurology 5% Primary Health Care – General Practice 4% Postgraduate Medical Trainee - Pediatric Medicine 4% Primary Health Care - Other 4% Tertiary Health Care - Pediatric Epileptologist 4% |
| Table 2: Individuals responsible for the development of EpilepsyDiagnosis.Org |
| Members of the ILAE's Commission on Classification and Terminology (2009-2013), and this Commission's Diagnostic Manual Taskforce: Ingrid Scheffer, Sameer Zuberi, Sam Berkovic, Pippo Capovilla, Helen Zhang, Doug Nordli, Jeff Buchalter, Lynette Sadleir, Anne Berg, Mary Connolly, Laura Guilhoto, Edouard Hirsch, Sam Wiebe, Christian Korff, Andrew Lux, Yoshimi Sogawa, Elaine Wirrell, Stephan Schuele, Kate Riney. |
| Members of the ILAE's Commission on Classification and Terminology (2013-2017) EpilepsyDiagnosis.Org and Syndromes Taskforce: Roberto Caraballo, Kate Riney, Norimichi Higurashi, Vivek Jain, Floor Jansen, Mike Kerr, Lieven Lagae, John Paul Leach, Ingrid Scheffer, Rima Nabbout, Elizabeth Thiele, Federico Vigevano, Khaled Zamel, Sameer Zuberi, Muhammad Salisu, Nerses Bebek. |
Read More
- Details
- ICNA
- News
- Hits: 948
Organelles and Neurometabolic Disease: a practical approach
This year pediatric neurologists from all over the world gather in Amsterdam for ICNC 2016. Amsterdam has a long tradition in research on neurometabolic disorders. Pediatric neurologists with an interest in metabolic disease should also consider the symposium “Organelles and Neurometabolic Disease: a practical approach”, April 30th, prior to the start of ICNC 2016.
Program is available here and registration is available through this link: https://amc.congrezzo.nl/satellite/registration
Contact: This email address is being protected from spambots. You need JavaScript enabled to view it.
Event: Satellite Symposium Organelles and Neurometabolic Disease- A Practical Approach
Date: April 30, 2016
Venue: Academic Medical Centre, Lecture Hall 5, Meibergdreef 9, 1105 AZ Amsterdam
Read More
- Details
- ICNA
- News
- Hits: 732
The U.S Centers for Disease Control have published interim guidelines evaluation and testing of infants born to mothers who may have been exposed to Zika virus during pregnancy. The guidelines suggest that pediatric health providers should work together with obstetric providers in order to identify infants whose mothers may have been exposed to Zika virus during pregnancy and fetal ultrasounds should be reviewed and maternal testing for Zika virus should be considered. Infants with microcephaly or intracranial calcifications born to women who traveled to or resided in an area with Zika virus transmission during pregnancy, and infants born to mothers with positive or inconclusive results of Zika virus infection, should undergoing Zika virus testing. If laboratory evidence of possible congenital Zika virus infection is found, those infants should undergo further clinical evaluation and follow-up. The only way to prevent congenital Zika virus infection is to prevent maternal infection, either by avoiding areas where Zika virus transmission is ongoing or strictly following steps to avoid mosquito bites.
Staples JE, Dziuban EJ, Fischer M, et al. Interim Guidelines for the Evaluation and Testing of Infants with Possible Congenital Zika Virus Infection — United States, 2016. MMWR Morb Mortal Wkly Rep. 2016; 65(Early Release):1–5. doi:10.15585/mmwr.mm6503e3er.
Read More
- Details
- ICNA
- News
- Hits: 795
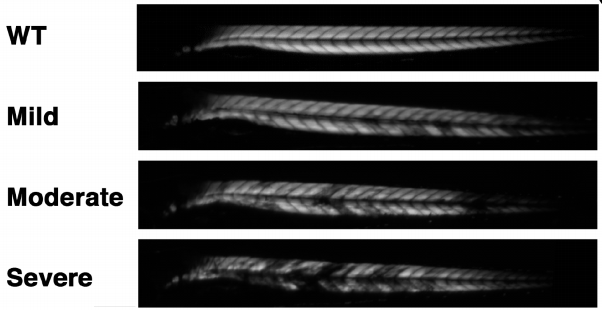
Professor Winder's group at the University of Sheffield investigating the cancer drug, dasatinib, a potent and specific Src tyrosine kinase inhibitor has shown that it decreases the levels of β-dystroglycan phosphorylation on tyrosine and to increase the relative levels of non-phosphorylated β-dystroglycan in dystrophic sapje zebrafish. Tyrosine phosphorylation and degradation of β-dystroglycan is a key event in the aetiology of Duchenne muscular dystrophy.
Dasatinib treatment resulted in the improved physical appearance of the sapje zebrafish musculature and increased swimming ability as measured by both duration and distance of swimming of dasatinib-treated fish compared with control animals. These findings show great promise for pharmacological agents that prevent the phosphorylation of β-dystroglycan on tyrosine and subsequent steps in the degradation pathway as therapeutic targets for the treatment of Duchenne muscular dystrophy.The results are published in the journal Human Molecular Genetics.
Since dasatinib is already cleared for clinical use in Leukemia, researchers are hopeful that progress can be made more quickly towards trialling the drug in humans as a treatment for DMD. It could be that by combining the drug with other treatments currently under development, their effectiveness could be improved even further. Experiments have already begun in mice models, with promising results.
Citation: Lipscomb L, Piggott RW, Emmerson T, Winder SJ (2015) Dasatinib as a treatment for Duchenne muscular dystrophy.Hum Mol Genet ():. DOI: 10.1093/hmg/ddv469 PMID: 26604135.
Cover image: Muscle birefringence images from wild type, mild, moderate and severely affected sapje fish.
Read More
- Details
- ICNA
- News
- Hits: 875