
Children from low income environments appear to have a higher risk of neurological impairment than those from more economically secure circumstances, according to researchers at the National Institutes of Health and other institutions. This neurological impairment appears to be distinct from the risk of cognitive and emotional delays known to accompany early-life poverty.
In most cases, the level of neurological impairment the researchers found would not be apparent to a casual observer. That level could, however, increase, the risk for childhood learning difficulties, attention deficit disorders and psychological conditions such as anxiety disorders and schizophrenia. “The size of the effect we saw was modest,” said the study’s senior author, Stephen Gilman, Sc.D., acting chief of the Health Behavior Branch at NIH’s Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD).
“However, the findings do indicate that an impoverished environment may pose a hazard for a child’s developing nervous system.” The study was published in the International Journal of Epidemiology. The researchers analyzed data from the Collaborative Perinatal Project (CPP), funded by NIH’s National Institute of Neurological Disorders and Stroke.
The current study was funded by NICHD and NIH’s National Institute of Mental Health. The CPP enrolled pregnant women from 1959 through 1966, obtaining health information on more than 50,000 pregnancies and the children resulting from them. Children in the study received comprehensive neurological examinations at birth, 4 months, 1 year and 7 years of age.
The physicians performing the examinations looked for obvious deformities, abnormalities in posture, motor skills, response to skin stimulation and muscle strength. The children also received evaluations of the autonomic nervous system — the part of the nervous system governing functions not under conscious control, such as breathing, heartbeat and digestion.
Based on interviews at the start of the study, the researchers classified the parents into three groups: those having a low, medium, or high likelihood of socioeconomic disadvantage based on such factors as educational level, income relative to the U.S. poverty level, occupation, employment status, and whether there were two parents living in the home.
When the researchers factored in the likelihood for pregnancy and birth complications—more common among women in poverty — they found little difference in neurological impairment at birth between the children, despite their parents’ socioeconomic disadvantage. However, beginning at age 4 months, the chance of having a neurological abnormality was higher in the most disadvantaged children (12.8 percent), compared to the least disadvantaged (9.3 percent). By age 7, the likelihood of a neurological abnormality increased to 20.2 percent among the most disadvantaged, compared to 13.5 percent among the least disadvantaged.
The greater frequency of pregnancy complications in the most disadvantaged group did not account for its higher percentage of neurological impairment. Although there have been advances in the techniques used to diagnose neurological impairment in the years since the data were collected, the study authors said that the diagnostic approaches used in the study are still effective for detecting neurological problems. Studies indicate that people living in poverty are at higher risk for substance abuse, anxiety, depression, and child abuse, and the authors theorize that these factors could explain the higher rates of neurological impairment their study found for children raised in impoverished environments.
The authors wrote that further research into how childhood poverty might contribute to neurological impairment could lead to ways to prevent neurological impairment from occurring. They added that the percentage of children living below the federal poverty threshold is higher today than it was when the CPP data were collected.
Researchers from the following institutions also participated in the study: Taipei City Hospital, Taiwan; Harvard School of Public Health, Boston; Lahey Hospital and Medical Center, Burlington, Massachusetts.; Brown University School of Public Health, Providence, Rhode Island.; Brigham and Women’s Hospital, Boston; Harvard Medical School and Boston Children’s Hospital, Boston; and Massachusetts General Hospital, Boston.
Reference:
Chin-Lun Hung G, Hahn J, Alamiri B, Buka SL, Goldstein JM, Laird N, Nelson CA, Smoller JW, Gilman SE. Socioeconomic disadvantage and neural development from infancy through early childhood. International Journal of Epidemiology, 2015
Source: National Institutes of Health (NIH)
Photo credit: Jose-Luis Olivares/MIT
Read More
- Details
- ICNA
- News
- Hits: 853

E-Rare-3 Call for Proposals 2016 for "Clinical research for new therapeutic uses of already existing molecules (repurposing) in rare diseases".
The eighth E-Rare joint call for funding multilateral research projects on rare diseases (JTC2016) will be open on the 7 December 2015. The following 14 countries intend to participate in this call: Austria, Canada (including Quebec), France, Germany, Hungary, Israel, Italy, Latvia, Poland, Portugal, Romania, Spain, Switzerland, Turkey.
The specific objective of this call is to promote the clinical and pre-clinical proof of concept for the potential application of medicinal products in rare indications either already marketed or having achieved a significant stage in the development process.
Two types of projects will be eligible for this call:
Type A: Preclinical studies to verify target engagement and to perform additional toxicity testing if necessary (for example in the case of paediatric indications where juvenile animal studies might be warranted) in a disease model for a maximum period of one year followed by the implementation of Phase 1b or Phase 2a clinical trials at the beginning of the second year of the project.
Type B: Milestone-driven Phase 2 clinical trials to demonstrate that the Agent modulates the target and has the potential to yield the desired clinical outcome in the proposed disease population for a period up to three years. Projects shall involve a group of rare diseases or a single rare disease following the European definition i.e. a disease affecting not more than five in 10.000 persons in the European Community, EC associated states and Canada.
The use of existing European health research infrastructures or initiatives is strongly encouraged when appropriate. The following European Research Infrastructures or Initiatives were identified as potentially useful for this kind of study: BBMRI; EATRIS; ECRIN; ELIXIR; EU-OPENSCREEN; INFRAFRONTIER; RD-Connect and European Medicines Agency
For more information, visit the E-Rare website http://erare.eu/
Read More
- Details
- ICNA
- News
- Hits: 953
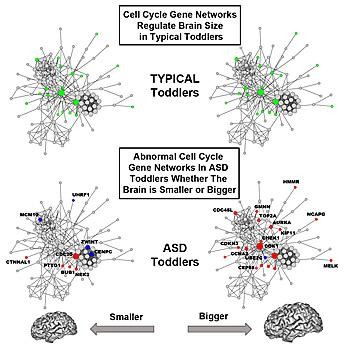
Further underscoring the prenatal origins of Autism Spectrum Disorder (ASD), researchers at University of California, San Diego School of Medicine describe for the first time how abnormal gene activity in cell cycle networks that are known to control brain cell production may underlie abnormal early brain growth in the disorder.
The findings are published online December 14 in Molecular Systems Biology.
"These findings identify common genomic defects that help explain why there are abnormal numbers of brain cells in autism, why the brain grows abnormally too large or too small in some ASD toddlers and how previously reported diverse gene mutations may, in fact, converge in their effects via common genomic pathways," said senior study author Eric Courchesne, PhD, professor of neurosciences and director of the Autism Center of Excellence at UC San Diego.
"This study, along with the work of others and our past findings on excess neurons and brain growth patterns, enable researchers to now trace the origins of early abnormal brain overgrowth in ASD back to prenatal disruption of networks governing brain cell numbers."
The proper regulation of brain cell numbers is one of earliest critical steps in fetal brain development. Courchesne and colleagues first discovered in 2001 that early brain overgrowth occurs in a substantial subset of ASD toddlers. In 2011, they reported that the young ASD prefrontal cortex, the region of the brain associated with social and communications skills, exhibited a 67 percent excess in cortical cells. In 2014, they described patches of disrupted cortical development in the brains of ASD children.
Rare gene mutations capable of increasing or decreasing cell number and brain size have been reported for tiny percentages of all ASD individuals, but for the vast majority of ASD children, the genomic defects behind abnormal brain overgrowth or undergrowth have remained unknown. The new study points to a common underlying defective functional genomic network - cell cycle - in living ASD toddlers, which plays a central role in fetal brain development.
Courchesne noted that several hundred different genes coordinate the cell cycle network in the fetal brain. "When they are active in the right way at the right time, they generate the correct number and type of brain cells that go to the cortex to create the normal brain cell layers and connections," he said. "Both genetic mutations and non-genetic factors can change how these genes work and cause abnormal cell numbers and cell types."
Attempts to better understand the genetic mechanisms underlying ASD are hindered by, among other things, the impossibility of direct brain gene expression measurement during critical periods of early development. However, first author Tiziano Pramparo, PhD, said blood and brain tissues highly express many of the same gene networks, particularly those related to cell cycle functions.
In the current study, researchers investigated the relationships between blood gene expression and brain size in 142 male toddlers, ages 1 to 4 years, 87 with a diagnosis of ASD and 55 controls.
In the control toddlers, researchers found that variation in brain size significantly correlated with cell cycle and protein folding gene networks, potentially impacting neuron number and synapse development. In ASD toddlers, correlations with brain size were strikingly disrupted due to considerable changes in the organization of cell cycle gene networks. Conversely, cell adhesion gene networks, which regulate the ability of cells to stick together to form multicellular tissues and communicate, showed an abnormal relationship with variation in brain size in ASD toddlers.
They also identified 23 candidate genes for brain maldevelopment, of which 4 are directly linked to genes frequently mutated in ASD. "These patterns of activity in these 23 genes relative to brain size in the ASD toddlers was very different from that in typical toddlers," observed Pramparo.
Pramparo said the new findings provide in vivo evidence for the involvement of cell cycle processes in ASD brain maldevelopment and significantly illuminate the complexities involved in early dysregulation and disruption of the developing ASD brain. Specifically, he noted:
They point to a convergence of findings related to mutation, gene expression and brain-related phenotypes in ASD, "which is key since ASD is very heterogeneous, both at the genetic and clinical level."
Many "high-confidence" ASD candidate genes are upstream and may control expression of other genes and networks with primary cell cycle functions.
Timing of the cell cycle appears to be critical. Shorter cell cycle periods may result in bigger brains and greater dysregulation. "The difference in smaller versus bigger may be due to genetic backgrounds, but also to non-genetic triggering events, especially when they occur during development."
Courchesne said the findings raise the possibility that future research could test whether genomic biomarkers of cell cycle disruption could be an early sign of ASD risk or might be indicators of clinical progression, severity and outcome.
"Our next studies will examine whether infants at risk for ASD who have the most deviant cell cycle expression patterns also have more abnormal early development of brain structure, connections and function," he said. "The findings do not directly suggest new therapies for children with ASD, but they raise the important question of whether the degree of genomic disruption might be related to treatment responsiveness."
Exploring this question, he said, along with how development of neural connectivity and functional patterns is altered in ASD brains, will be addressed in future research.
Reference:
http://dx.doi.org/10.15252/msb.20156108
Cover image:
Gene function in cell cycle networks regulating brain size is normal in typical toddlers (green), while it is abnormal in ASD toddlers (blue, red) for both smaller and bigger brains. Bigger ASD brains are more severely affected than smaller brains.
Read More
- Details
- ICNA
- News
- Hits: 1025
Douglas E. Crompton and colleagues from Northern Health in Melbourne looked at the demographic and clinical information on SUDEP cases from 2 major centers in Australia and performed whole exome sequencing to identify rare genetic variants in these patients.
They compared genes with an increased prevalence of rare pathogenic variants in SUDEP patients with 2936 control exomes. The study findings were, presented at the 2015 American Epilepsy Society Annual Meeting in Philadelphia.
For the present study they analysed whole exome sequences from 62 SUDEP cases. The mean age at epilepsy onset was 10.5 years, while the mean age of SUDEP was 28 years. The male to female ratio was 1.2:1
The cardiac sudden related death gene NOSAP1, the long QT syndrome gene KCNH2 and the focal epilepsy gene DEPDC5 were ranked among the top 30 genes of the entire exome interrogated (n=15,294 genes).
Six SUDEP cases (10%) had mutations in genes commonly responsible for the cardiac arrhythmia disorder long QT syndrome, while eight cases (13%) had mutations in other cardiac arrhythmia genes.
Fifteen cases (24%) had sixteen mutations in known epilepsy genes, including six individuals with mutations in the focal epilepsy gene DEPDC5.
According to the study authors the discovery of mutations known to cause cardiac sudden death in nearly one quarter of their SUDEP cohort suggests that cardiac dysrhythmia may contribute to pathophysiology in a proportion of SUDEP cases.
The researchers further suggest that these findings raise the possibility that SUDEP might be preventable in some cases by restricting the use of anti-epileptic drugs known to prolong QT interval, and in selected cases, by recommending beta-blockers, pacemakers, or implantable cardioverter-defibrillators.
The high prevalence of DEPDC5 mutations in their cohort might signal a particular SUDEP predisposition in patients with these mutations. Although no single gene reached exome-wide significance in their study cohort the collapsing analysis (unconstrained by prior hypothesis as to which genes are relevant) identifies genes which are biologically plausible as contributors to SUDEP.
Analyses of larger SUDEP cohorts hold promise for identifying hitherto unsuspected genes as contributors in SUDEP pathophysiology, providing vital insights into pathophysiology and ultimately prevention of SUDEP.
Reference:
Crompton DE, Bagnall R, Petrovski S, et al. Abstract 3.339|C.01. Identifying genetic variants underlying sudden unexpected death in epilepsy (SUDEP). Presented at: American Epilepsy Society Annual Meeting; Dec. 4-8, 2015; Philadelphia.
Read More
- Details
- ICNA
- News
- Hits: 933
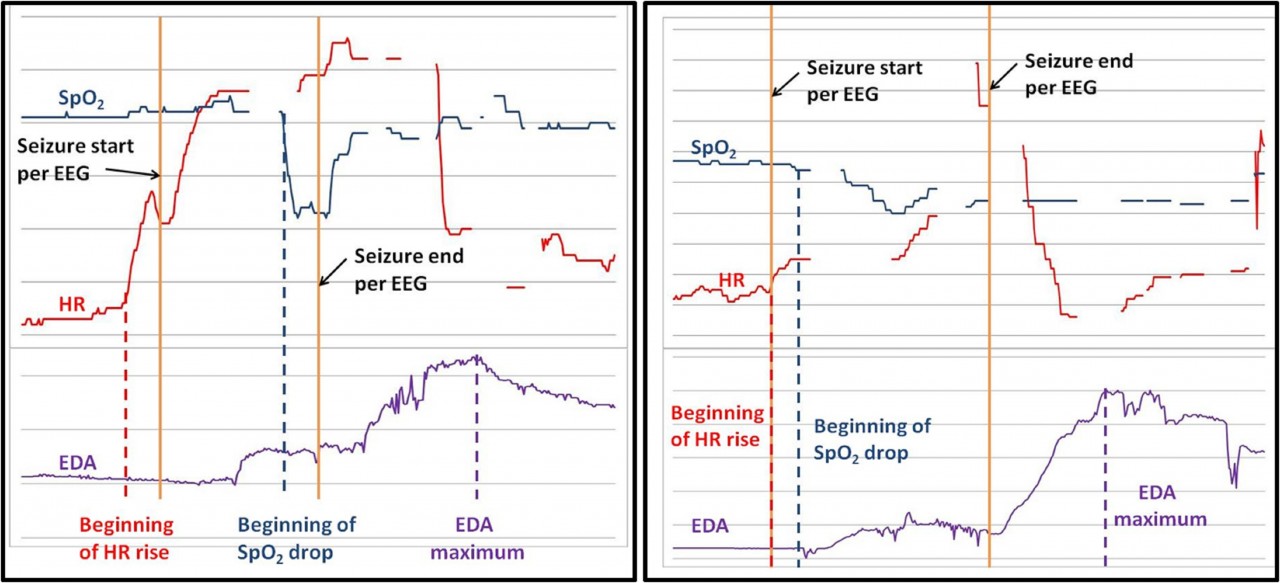
Seizure detection using multiple biosignals more accurate than detection by heart rate changes alone
According to data presented at the 2015 American Epilepsy Society Annual Meeting in Philadelphia seizure detection using multiple extracerebral biosignals specifically [HR], [SpO2] and electrodermal activity [EDA] results in improved accuracy compared with heart rate alone.
Using 2 commercially available wrist-worn devices, Cogan and colleagues gathered HR, arterial oxygenation (SpO2), and EDA data from 20 patients electively admitted to an epilepsy-monitoring unit. Of these patients, 11 provided HR, SpO2, and EDA data on 24 seizures during 355 hours of data collection.
In all 24 of the captured seizures HR increased by 15% or more. In 20 of these seizures, an SpO2 drop of 5% or more was seen immediately following the HR rise. This combination of HR↑ + SpO2↓ is less likely to be seen in non-seizure situations than an increase in HR alone.
Two complex partial seizures (CPS) resulted in less pronounced SpO2 disturbances which might require capture with more advanced (e.g. wavelet based) signal processing. SpO2 was undisturbed in only two CPS.
Of the 20 seizures with HR↑ + SpO2↓ combinations, 12 showed strong EDA responses (increases of at least 60% and 1μS), 6 showed weak EDA responses (increases of at least 60% but less than 1μS), and 2 showed no EDA response.
The team plans to test the hypothesis that EDA responses to focal seizures are contralateral by collecting data from both wrists [Poh, Ming-Zher, “Continuous Assessment of Epileptic Seizures with Wrist-worn Biosensors,” PhD diss., Massachusetts Institute of Technology, 2011]. If the hypothesis is proved correct stronger EDA responses will be seen on the wrist opposite the seizure focus.
The researchers also hope to present a pattern recognition algorithm later this year which further improves seizure detection accuracy for these patients and adopt device models which can be worn on the wrist. Data obtained from such devices will also help understand the causes and early signs of sudden unexpected death in patients with epilepsy.
Reference:
Cogan DL, Nourani M, Harvey J, Nagaraddi V. Abstract 3.084. Seizure detection by multi extracerebral biosignal analysis. Presented at: American Epilepsy Society Annual Meeting; Dec. 4-8, 2015; Philadelphia.
Cover image: illustrates multi extracerebral biosignal responses to a Complex Partial Seizure (left) and a Generalized Tonic Clonic Seizure (right).
Disclaimer: The International Child Neurology Association (ICNA) or the American Epilepsy Society (AES) do not endorse or recommend any particular device or technology.
Read More
- Details
- ICNA
- News
- Hits: 876