
- Details
- ICNA
- News
- Hits: 1238
It is a great pleasure to welcome you to submit proposals for symposia for the 15th International Child Neurology Congress that will take place in the enchanting city of Mumbai, India, November 15-18, 2018. The theme for the Congress is "Protecting the developing brain and preventing disability". The ACON is the local organizer and host of the Congress, in which Child Neurologists from all over the world will meet at the Grand Hyatt Hotel in downtown Mumbai.
We invite you to submit proposals for Symposia. All proposals must be submitted through this website in the period from May 08, 2017 to September 30, 2017. Three to four symposia will be held concurrently each morning of the four day Congress.

Each symposium will be 2 hours in duration and would be expected to have a total of 3-4 speakers drawn from diverse geographic origins. A symposium is meant to be a focused session on a common theme. The format usually involves introduction and background to the discussion by the symposium chair, followed by talks from participants. The viewpoints may differ and there would be interaction between audience and participants. The symposium would end with a summary overview of the session by the chair. Proposals are invited from a proposed Chair who will be the principal organiser of the symposium, and who may have one other international co-organiser.
Chairs are encouraged to submit the overall proposal before September 30, 2017 to allow enough time for individual participants to complete their portion of the proposal once the initial proposal is accepted. The submitted symposia proposals will be evaluated by the SPC and the selected Symposia will be announced in December 2017. A list of possible topics can be seen here, which is not meant to be restrictive but is included to stimulate thinking about areas of great interest.
The Program Committee includes distinguished representatives from ICNA (The International Child Neurology Association), AOCNA (Asian Oceanian Child Neurology Association) and AOCN (Association of Child Neurologists, India), and plans to develop a high quality scientific program for ICNC 2018 that reflects our organizations' global interests as well as the latest scientific and clinical advances in Child Neurology.
We hope that you accept our invitation to submit a Symposium proposal to the SPC of the ICNC 2018. If you have any questions, please, do not hesitate to ask.
We also seek your suggestions for topics and speakers for workshops and meet the expert sessions. Workshops involve a skill building session incorporating theoretical knowledge and practical approaches to further understanding and clinical skills in a particular area. Meet the expert sessions usually include 1 to 2 presenters who are chosen for their expertise in a particular area of interest. The sessions are for interaction between the presenters and the audience. Typically involves prolonged discussion on audience questions.
Please send any suggestions you may have for workshops or meet the speaker sessions directly to me at any time.

Chair, Scientific Program Committee
Professor Neurology and Pediatrics
Kennedy Krieger Institute and
Johns Hopkins University School of Medicine
This email address is being protected from spambots. You need JavaScript enabled to view it.
Read More
- Details
- ICNA
- News
- Hits: 1142

Prof Lieven Lagae, ICNA Executive Board Member & Director of the Childhood Epilepsy Program at the KUL University Hospitals is the recipient of "Cures within Research" 5th Annual Global Health Repurposing Awards. Prof Lagae has been awarded the new Industry Patient Impact Clinical Award.
Award recipients will be recognized at the Global Health Repurposing Awards on June 27, 2017 at the 6th Annual Global Health Drug Repositioning and Repurposing Conference in Chicago, IL.
We are very proud to acknowledge the achievements of our fellow ICNA Executive Board member, Dr. Lieven Lagae of KUL University Hospitals in Belgium, for his "Cures Within Reach Industry Patient Impact Clinical Award 2017" which has been given in recognition of his pioneering research and world-wide efforts to repurpose the drug fenfluramine for Dravet syndrome patients.
Dravet syndrome is a rare, catastrophic, lifelong form of epilepsy that begins in the first year of life with frequent and/or prolonged seizures. Previously known as Severe Myoclonic Epilepsy of Infancy (SMEI), it affects 1:15,700 individuals, 80% of whom have a mutation in their SCN1A gene.
Fenfluramine is an amphetamine-like drug that has been used in the past as a part of antiobesity treatments. Because of the possible cardiac adverse effects (valve thickening, pulmonary hypertension) associated with use of fenfluramine, it was withdrawn from the market in 2001. Dr. Lagae has doggedly fought to bring Dravet patients access to this drug, and is leading a worldwide effort to secure regulatory approval for fenfluramine so that it will help stop these debilitating seizures.
In Belgium, a Royal Decree permitted examination of the potential anticonvulsive effects of fenfluramine in a clinical trial consisting of a small group of patients diagnosed with Dravet syndrome.
The results of a prospective, open-label study by Prof Lage and colleagues assessing the safety and effectiveness of low-dose fenfluramine in a new cohort of Nine patients (aged 1.2-29.8 years) with Dravet syndrome has been recently published in the European Journal of Neurology in February 2017. In this study following a 3-month baseline period, fenfluramine was added to each patient's current antiepileptic drug regimen at a dose of 0.25-1.0 mg/kg/day (max. 20 mg/day) for a median duration of 1.5 (range, 0.3-5.1) years.
All patients demonstrated a reduction in seizure frequency during the treatment period with a median reduction of 75% (range, 28-100%). Seven patients (78%) experienced a ≥50% reduction in major motor seizure frequency. The most common adverse events were somnolence (n = 5) and anorexia (n = 4). No evidence of cardiac valvulopathy or pulmonary hypertension was observed.
The effectiveness and safety of low-dose fenfluramine as an add-on therapy for Dravet syndrome in this new prospective cohort supported previous findings by the same researchers in smaller observational studies.
Cures Within Reach
Cures Within Reach, is a leading global nonprofit focused on repurposing research as a fast track to saving patient lives. Cures Within Reach (http://www.cureswithinreach.org) works to catalyze repurposing research to quickly and affordably improve patient lives. We accomplish this by connecting funders with researchers to jumpstart repurposing research clinical trials, by providing collaboration tools so repurposing stakeholders can work together more easily, and by pioneering alternative finance engines and incentives for repurposing research. Cures Within Reach's repurposing research projects have generated over a dozen "new" treatments making patient impact through off-label use in clinical practice or through a commercialization track.
Further reading
Low-dose fenfluramine significantly reduces seizure frequency in Dravet syndrome: a prospective study of a new cohort of patients.Schoonjans A, Paelinck BP, Marchau F, Gunning B, Gammaitoni A, Galer BS, Lagae L, Ceulemans B.Eur J Neurol. 2017 Feb;24(2):309-314. doi: 10.1111/ene.13195.
Five-year extended follow-up status of 10 patients with Dravet syndrome treated with fenfluramine.Ceulemans B, Schoonjans AS, Marchau F, Paelinck BP, Lagae L.Epilepsia. 2016 Jul;57(7):e129-34. doi: 10.1111/epi.13407. Erratum in: Epilepsia. 2017 Mar;58(3):509-510.
Low-dose fenfluramine in the treatment of neurologic disorders: experience in Dravet syndrome.Schoonjans AS, Lagae L, Ceulemans B.Ther Adv Neurol Disord. 2015 Nov;8(6):328-38. doi: 10.1177/1756285615607726.

Read More
- Details
- ICNA
- News
- Hits: 1199
A randomized, double-blinded trial has shown that pitolisant, a drug that targets a specific histamine receptor, can reduce cataplexy, a very disabling symptom of narcolepsy. Cataplexy is a sudden and uncontrollable muscle weakness or paralysis that comes on during the day and is often triggered by a strong emotion, such as excitement or laughter. The loss of muscle tone in cataplexy occurs because of the inability to regulate sleep and awake states -- meaning that elements of each can overlap. During normal rapid eye movement (REM) sleep, there is a natural loss of muscle tone. In the case of cataplexy, that characteristic of REM sleep occurs suddenly during the day, causing weakness or full paralysis, even as the person remains awake during the episode.
The study is published in the January 24 online edition of the Lancet Neurology. According to the lead author Jean-Charles Schwartz, PhD "If these data are confirmed in long-term studies, this drug could be useful for narcolepsy patients with or without cataplexy and those with less severe sleep disturbances,". Pitolisant is a histamine H3 receptor inverse agonist. Blocking the receptor with pitolisant activates histamine release in the brain and increases wakefulness and reduces cataplexy events.
Currently US Food and Drug Administration (FDA) approved therapies for narcolepsy which alter dopamine transmission and other off-label drugs used that act on serotonin and norepinephrine are not effective for treating cataplexy, The only FDA drug available for cataplexy is sodium oxybate (Xyrem), a short-acting liquid drug that must be swallowed twice during the night. The researchers Dr. Schwartz and Jeanne Marie Lecomte, PhD, started a biotechnology company, Bioprojet Pharma, and are now preparing an application to seek FDA approval of pitolisant.
Study
For this randomised, double-blind, placebo-controlled trial patients with narcolepsy were recruited from 16 sleep centres in nine countries (Bulgaria, Czech Republic, Hungary, Macedonia, Poland, Russia, Serbia, Turkey, and Ukraine). Patients were eligible if they were aged 18 years or older, diagnosed with narcolepsy with cataplexy according to version two of the International Classification of Sleep Disorders criteria, experienced at least three cataplexies per week, and had excessive daytime sleepiness (defined as an Epworth Sleepiness Scale score ≥12).
They used a computer-generated sequence via an interactive web response system to randomly assign patients to receive either pitolisant or placebo once per day (1:1 ratio). Randomisation was done in blocks of four. Participants and investigators were masked to treatment allocation.
Treatment lasted for 7 weeks: 3 weeks of flexible dosing decided by investigators according to efficacy and tolerance (5 mg, 10 mg, or 20 mg oral pitolisant), followed by 4 weeks of stable dosing (5 mg, 10 mg, 20 mg, or 40 mg). The primary endpoint was the change in the average number of cataplexy attacks per week as recorded in patient diaries (weekly cataplexy rate [WCR]) between the 2 weeks of baseline and the 4 weeks of stable dosing period. Analysis was by intention to treat.
117 patients were screened, 106 of whom were randomly assigned to treatment (54 to pitolisant and 52 to placebo) and, after dropout, 54 patients from the pitolisant group and 51 from the placebo group were included in the intention-to-treat analysis.
The WCR during the stable dosing period compared with baseline was decreased by 75% (WCRfinal=2·27; WCRbaseline=9·15; WCRfinal/baseline=0·25) in patients who received pitolisant and 38% (WCRfinal=4·52; WCRbaseline=7·31; WCRfinal/baseline=0·62) in patients who received placebo (rate ratio 0·512; 95% CI 0·43–0·60, p<0·0001).
Treatment-related adverse events were significantly more common in the pitolisant group than in the placebo group (15 [28%] of 54 vs 6 [12%] of 51; p=0·048).
There were no serious adverse events, but one case of severe nausea in the pitolisant group. The most frequent adverse events in the pitolisant group (headache, irritability, anxiety, and nausea) were mild or moderate except one case of severe nausea. No withdrawal syndrome was detected following pitolisant treatment; one case was detected in the placebo group.
The researchers concluded that pitolisant was well tolerated and efficacious in reducing cataplexy. If confirmed in long-term studies, pitolisant might constitute a useful first-line therapy for cataplexy in patients with narcolepsy, for whom there are currently few therapeutic options.
Histamine and Sleep
Dr. Schwartz 's lab discovered histamine's role as a neurotransmitter in 1975 and subsequently Dr. Schwartz identified the H3 receptor. He went on to describe its critical role in the sleep-wake cycle, and designed a drug that singularly targets these receptors and showed that blocking these receptors would work as a treatment for narcolepsy.
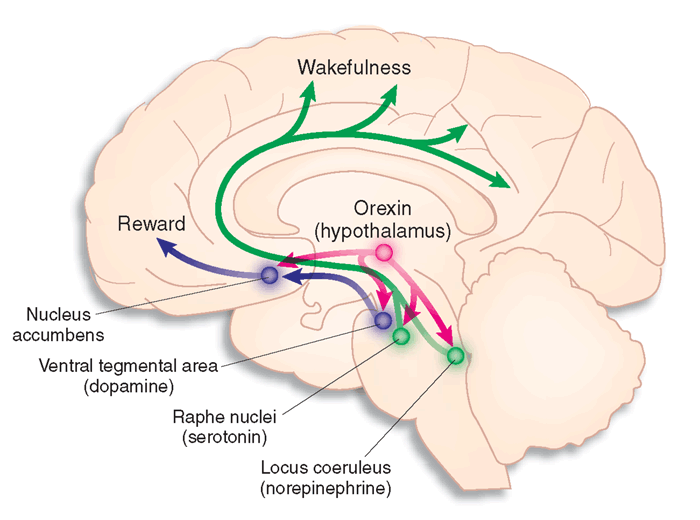
The studies that followed led to pitolisant's approval in Europe in 2015. Histamine is released from a group of only 2,000 neurons, specifically in the tuberomammillary nucleus of the hypothalamus that innervates the brainstem and up into the basal forebrain. Cataplexy, an autoimmune disease, leads to loss of hypocretin (also known as orexin) neurons, which are located in a nearby hypothalamic pathway responsible for regulating the activity of wake-promoting monoaminergic systems, like histamine.
Citation
Szakacs Z, Dauvilliers Y, Mikhaylov V, et alfor the HARMONY-CTP Study Group. Safety and efficacy of pitolisant on cataplexy in patients with narcolepsy: A randomized, double-blind, placebo-controlled trial http://http://www.thelancet.com/journals/laneur/article/PIIS1474-4422(16)30333-7/abstract. Lancet Neurol 2017; Epub 2017 Jan 24.
Dauvilliers Y, Bassetti C, Lammers GJ, Schwartz JC. Pitolisant versus placebo or modafinil in patients with narcolepsy: A double-blind, randomized trial http://www.thelancet.com/journals/laneur/article/PIIS1474-4422(13)70225-4/abstract. Lancet Neurol 2013;12(11):1068–1075.
Cover: Orexin projections from hypothalamus to the brain. Image source: http://www.nature.com/nm/journal/v13/n2/images/nm0207-126-F1.gif
{kind=link}
Read More
- Details
- ICNA
- News
- Hits: 2096
Researchers have linked Nodding syndrome, a devastating form of pediatric epilepsy found in specific areas of east Africa, to a parasitic worm that can cause river blindness. The study, published in Science Translational Medicine, suggests that the mysterious neurological disease may be caused by an autoimmune response to the parasitic proteins.
"This study identifies a cause of Nodding syndrome. But more broadly, these findings provide a novel perspective on epilepsy and suggest that some forms of this neurological disorder may be autoimmune in nature," said Avindra Nath, M.D., clinical director of the NIH's National Institute of Neurological Disorders and Stroke (NINDS).
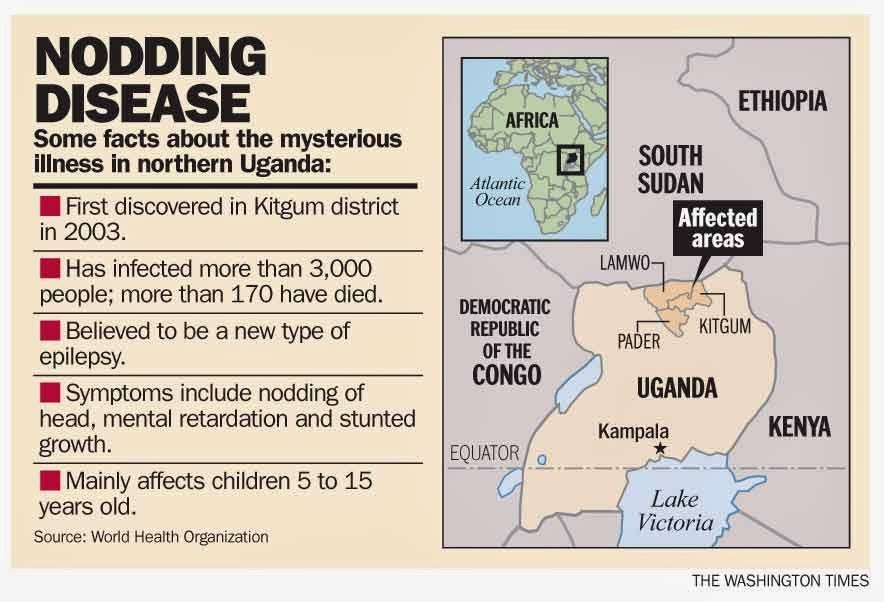
Nodding syndrome is a form of epilepsy that occurs in children between the ages of 5 and 16 who live in distinct regions of Tanzania, Uganda and the Republic of South Sudan. It is characterized by head nodding, seizures, severe cognitive deterioration and stunted growth. Nodding syndrome may lead to malnutrition and patients have died through seizure-associated traumas such as fatal burns and drowning.
Many studies have reported an association between Nodding syndrome and Onchocerca volvulus, a parasitic worm that can also cause river blindness. The worm is spread by black flies in specific geographic areas, where clusters of Nodding syndrome have been observed. However, it was unclear whether the worm caused this neurological disorder.
In this study, Nath and his colleagues compared serum samples from patients with Nodding syndrome and healthy controls who all lived in the same village in Uganda.
The results showed high levels of antibodies to leiomodin-1 in the samples obtained from patients. In addition, antibody to leiomodin-1 was also present in cerebrospinal fluid of patients with Nodding syndrome. Previous studies have shown leiomodin-1 is found in muscles, but this was the first time researchers saw it in the nervous system.
To confirm that finding, Nath's team examined brain tissue and found leiomodin-1 inside brain cells, notably in regions associated with symptoms of Nodding syndrome. Furthermore, when healthy neurons in a dish were treated with serum from the patients and antibodies against leiomodin-1, they did not survive, but removing the antibodies increased brain cell survival.
In addition, Nath and his group found that antibodies that bind to leiomodin-1 also attach to proteins from Onchocerca volvulus. Structurally, leiomodin-1 was shown to be very similar to specific proteins from that parasite.
The results of this study suggest that Nodding syndrome may be an autoimmune disease, in which the immune system incorrectly attacks the body's own proteins. According to the researchers, the immune system creates antibodies to fight off the parasite following infection with Onchocerca volvulus. However, those antibodies also bind to leiomodin-1, so the immune system - incorrectly - will attack brain cells that contain that protein, which can result in symptoms of Nodding syndrome.
"The findings also suggest that therapies targeting the immune system may be effective treatments against this disorder and possibly other forms of epilepsy," said Nath. "Another huge implication of this study is that exterminating black flies and getting rid of the parasite should stop the disorder from occurring."
More research is needed to learn about the role of leiomodin-1 in healthy people as well as in individuals with epilepsy. For example, one-third of controls also had leiomodin-1 antibodies, but it is unclear whether these individuals may eventually develop Nodding syndrome.
Nath's team is currently developing an animal model of Nodding syndrome to further study the disease and test potential therapies.
This work was supported by the NIH Intramural Program.
Article: Nodding syndrome may be an autoimmune reaction to the parasitic worm Onchocerca volvulus, Tory P. Johnson et al., Science Translational Medicine, doi: 10.1126/scitranslmed.aaf6953, published 15 February 2017.
Read More
- Details
- ICNA
- News
- Hits: 1140
Following a prolonged epileptic seizures neural connections of the brain may be rewired in an incorrect way. This may result in seizures that are difficult to control with medication. Mechanisms underlying this phenomenon are not entirely known, which makes current therapies ineffective in some patients.
In a study, published in the Annals of Neurology, researchers at the Neuroscience Center of the University of Helsinki have shown a role for gamma-aminobutyric acid (GABA), a main neurotransmitter in the brain, in the glutamatergic network rewiring in the brain. Rewiring of excitatory glutamatergic neuronal circuits is a major abnormality in epilepsy.
After a prolonged convulsive seizure, instead of the usual inhibitory effect of the transmitter, GABA accelerates brain activity. This, in turn, creates new, harmful neural connections.
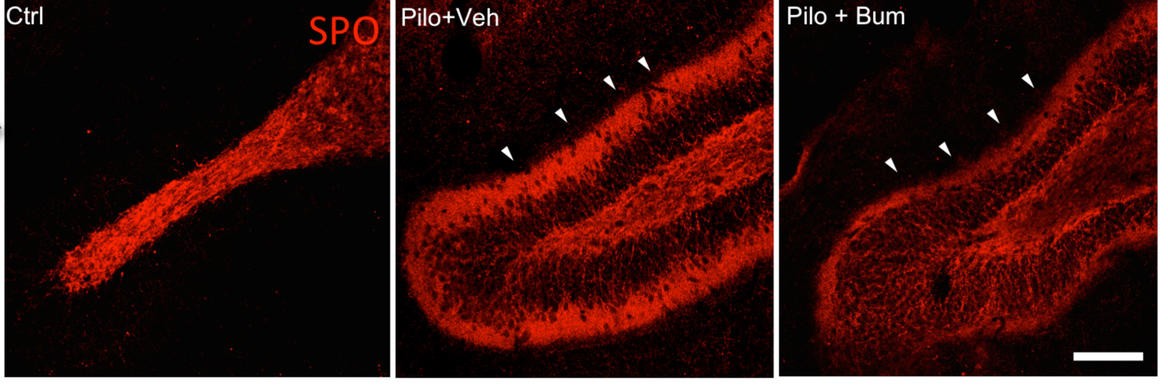
Using a rat model of epilepsy they also found that accelerating effect of GABA was blocked for three days with a drug called bumetanide given soon after a seizure. The number of harmful connections detected in the brain was significantly lower two months later. Although there is extensive literature on the role of bumetanide in the acute treatment of seizures, this study has shown for the first time, that bumetanide has a long term effect on the neural network structure of the brain.
Citation
Nazim Kourdougli, Christophe Pellegrino, Juho-Matti Renko, Stanislav Khirug, Geneviève Chazal, Tiina-Kaisa Kukko-Lukjanov, Sari E. Lauri, Jean-Luc Gaiarsa, Liang Zhou, Angélique Peret, Eero Castrén, Raimo K. Tuominen, Valérie Crépel, Claudio Rivera. Depolarizing γ-aminobutyric acid contributes to glutamatergic network rewiring in epilepsy. Annals of Neurology, 2017; 81 (2): 251 DOI: 10.1002/ana.24870
Cover: The amount of harmful nerve connections is significantly lower (far right, arrows) when treating with bumetanide after a prolonged convulsive seizure. Credit: Nazin Kourdougli
Read More