- News
- Eteplirsen trial results announced- potential breakthrough in DMD treatment
Eteplirsen trial results announced- potential breakthrough in DMD treatment

The 48-week results from the ongoing Phase IIb clinical trial of eteplirsen for the treatment of Duchenne muscular dystrophy (DMD)has been announced. Eteplirsen is an exon-skipping compound that addresses one of the underlying genetic defects in Duchenne muscular dystrophy.
At 48 weeks eteplirsen demonstrated a significant and unprecedented clinical benefit on the primary clinical outcome measure, the 6-minute walk test, and met the primary efficacy endpoint of the study, an increase in novel dystrophin. The results represent the potential medical breakthrough that eteplirsen represents for the treatment of DMD.
Although only about 13% of boys with Duchenne muscular dystrophy have the specific mutation targeted by eteplirsen, the implications for all Duchenne muscular dystrophy patients with related genetic mutations are clearly evident.
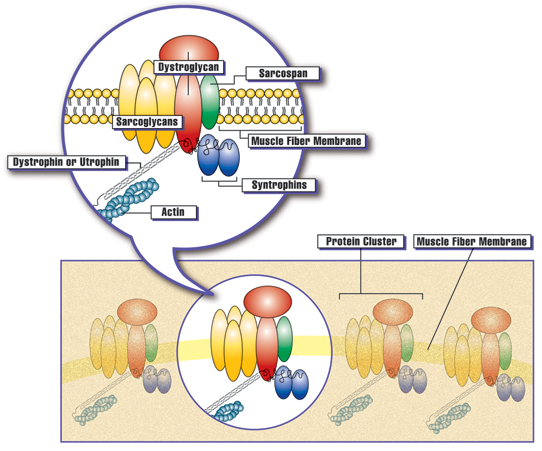 Clusters of proteins are spaced at intervals along the membrane of each muscle fiber. The presence of dystrophin, or the related protein utrophin, appears to be necessary for the other proteins in the cluster to assemble at the membrane. Courtesy: Muscular Dystrophy Association
Clusters of proteins are spaced at intervals along the membrane of each muscle fiber. The presence of dystrophin, or the related protein utrophin, appears to be necessary for the other proteins in the cluster to assemble at the membrane. Courtesy: Muscular Dystrophy Association
Trial Results
Eteplirsen administered once weekly at either 30mg/kg or 50mg/kg for 48 weeks (n=8) resulted in a statistically significant increase in dystrophin-positive fibers of 47.0% of normal.
The placebo/delayed treatment cohort, which received 24 weeks of eteplirsen at either 30mg/kg or 50mg/kg following 24 weeks of placebo (n=4), produced a statistically significant increase in dystrophin-positive fibers of 38.3% of normal.
In addition, eteplirsen administered once weekly at 50mg/kg over 48 weeks resulted in a 89.4 meter benefit compared to patients in our placebo/delayed treatment arm, those patients who received placebo for 24 weeks followed by 24 weeks of treatment with eteplirsen in the open-label extension of the study.
Upon evaluating all study participants through 48 weeks, no treatment-related adverse events which further demonstrate the highly favorable safety profile of eteplirsen.
An abstract describing the results from this Phase IIb extension study has been accepted as part of the World Muscle Society (WMS) Congress's Late-Breaking Science program in Perth, Australia during October 9 to October 13, 2012.
Principal investigator, Jerry R. Mendell, M.D. of Nationwide Children's Hospital, will present the data "Results at 48 Weeks of a Phase IIb Extension Study of the Exon-Skipping Drug Eteplirsen in Patients with Duchenne muscular dystrophy (DMD)" at the World Muscle Society (WMS) Congress's Late-Breaking Science program in Perth, Australia on October 13 at 4:00 p.m. WST UTC +8 hours/4:00 a.m. EDT.
Summary of Dystrophin: Eteplirsen-Treated Patients in All Dose Groups through Week 48*
| Treatment Arm | Mean Change from Baseline in % Dystrophin-Positive Fibers | p-value | |||
| Eterplirsen (both doses): 48 wks of Tx (n=8) | 47.0 | ≤0.001 | |||
| Eteplirsen 50 mg/kg (n=4) | 41.7 | ≤0.008 | |||
| Eteplirsen 30 mg/kg (n=4) | 52.1 | ≤0.001 | |||
| Placebo/Delayed Tx: 24 wks of Tx (n=4) | 38.3 | ≤0.009 | |||
| Placebo/50 mg/kg Delayed-Tx (n=2) | 42.9 | ns | |||
| Placebo/30 mg/kg Delayed-Tx (n=2) | 34.2 | ns | |||
* Values based on Immunofluorescence using anti-dystrophin antibody MANDYS106
Modified Intent-to-Treat (mITT)
The 6MWT results were further analyzed using the mITT population which excluded two patients who were randomized to the 30 mg/kg weekly eteplirsen cohort who showed signs of rapid disease progression within weeks after enrollment and were unable to perform measures of ambulation beyond 24 weeks. This mITT population consisted of 10 patients (4 eteplirsen-treated patients receiving 50 mg/kg weekly, 2 eteplirsen-treated patients receiving 30 mg/kg weekly, and 4 placebo/delayed-treatment patients).
Summary of 6MWT: Eteplirsen versus Placebo/Delayed-Treatment to Week 48*
| Treatment Arm | Mean Change from Baseline in 6MWT (meters) | Estimated Treatment Effect (Eteplirsen minus Placebo/Delayed-Tx) | p-value | |||
| Placebo/Delayed-Tx (n=4) | -60.3 | |||||
| Eteplirsen 50 mg/kg (n=4) | +27.1 | 87.4 m | ≤0.001 | |||
| Eteplirsen Both Doses (n=6) | +7.3 | 67.3 m | ≤0.001 | |||
| Eteplirsen 30 mg/kg (n=2) | -31.5 | 28.8 m | ns | |||
*Note: Analysis based on Mixed Model Repeated Measures test
Summary of Additional Sub-Group Analyses at Week 48*
| Subset | Mean 6MWT Change from Baseline (meters) | Estimated Treatment Benefit (Eteplirsen minus Placebo/delayed-Tx) | p-value | |||
| Placebo/delayed Tx: < 9.5 yrs at baseline (n=2; mean=7.6 yrs) |
-42.3 | 58.9 m | ≤0.038 | |||
| Eteplirsen: < 9.5 yrs at baseline (n=3; mean=8.4 yrs) |
+16.5 | |||||
| Placebo/delayed Tx: ≥9.5 yrs at baseline (n=2; mean=10.1 yrs) |
-63.5 | 52.1 m | ns | |||
| Eteplirsen: ≥9.5 yrs at baseline (n=3; mean=10.4 yrs) |
-11.3 | |||||
| Placebo/delayed Tx: Higher 6MWT baseline (n=2; mean=422m) |
-53.5 | 93.8 m | ≤0.001 | |||
| Eteplirsen: Higher 6MWT baseline (n=3; mean=424m) |
+40.3 | |||||
| Placebo/delayed Tx: Lower 6MWT baseline (n=2; mean=367m) |
-65.8 | 39.6 m | ns | |||
| Eteplirsen: Lower 6MWT baseline (n=3; mean=375m) |
-26.2 | |||||
| Placebo/delayed Tx: Genotype 49-50 deletion (n=3; age mean=9.2 yrs, 6MWT BL mean=397m) |
-69.0 | 83.4 m | ≤0.001 | |||
| Eteplirsen: Genotype 49-50 deletion (n=2; age mean=9.1 yrs, 6MWT BL mean=383m) |
+14.4 | |||||
* Note: Analysis based on Mixed Model Repeated Measures test
About Study 201 and Study 202 (Phase IIb Eteplirsen Study)
Study 4658-US-201 was conducted at Nationwide Children's Hospital in Columbus, Ohio. Twelve boys meeting the inclusion criteria being between 7 and 13 years of age with appropriate deletions of the dystrophin gene that confirm eligibility for treatment with an exon-51 skipping drug, received double-blind IV infusions of placebo (n=4), 30 mg/kg of eteplirsen (n=4), or 50 mg/kg of eteplirsen once weekly for 24 weeks (n=4). Muscle biopsies for evaluation of dystrophin were obtained at baseline for all subjects, and after 12 weeks for patients in the 50 mg/kg cohort and after 24 weeks for patients in the 30 mg/kg cohort. Two placebo patients were randomized to the 30 mg/kg cohort and two placebo patients were randomized to the 50 mg/kg cohort. This study design allowed to investigate the relationship of dose and duration of eteplirsen treatment on the production of dystrophin over the course of the 24-week study.
Study 4658-US-202 is the extension study to 201 and continues to assess the long-term safety and efficacy of open-label eteplirsen. The four placebo patients were rolled over to open-label eteplirsen at week 24, with six patients on 30 mgs/kg, and six patients on 50 mgs/kg. Third biopsies occurred at 48 weeks in the original study 201 treated patients, and at 24 weeks, the same time point, in the original placebo patients. 6MWT was performed at 32 weeks, 36 weeks, 48 weeks and will continue to be performed every 12 weeks going forward.
About Dystrophin
Dystrophin, a large structural protein, is critical to the stability of myofiber membranes in skeletal, diaphragmatic and cardiac muscle, protecting muscle fibers from contraction-induced damage. Loss of functional dystrophin destabilizes the dystroglycan protein complex, impairing its localization to the muscle membrane, and compromising the integrity of the membrane structure. The absence of functional dystrophin results in muscle membrane breakdown with muscle fibers being replaced by adipose and fibrotic tissue.
Eteplirsen
Eteplirsen uses phosphorodiamidate morpholino oligomer (PMO)-based chemistry and proprietary exon-skipping technology to skip exon 51 of the dystrophin gene enabling the repair of specific genetic mutations that affect approximately 13 percent of the total DMD population. By skipping exon 51, eteplirsen may restore the gene's ability to make a shorter, but still functional, form of dystrophin from messenger RNA, or mRNA. Promoting the synthesis of a truncated dystrophin protein is intended to improve, stabilize or significantly slow the disease process and prolong and improve the quality of life for patients with DMD.
About the 6-Minute Walk Test
The 6-minute walk test (6MWT) was developed as an integrated assessment of cardiac, respiratory, circulatory, and muscular capacity (American Thoracic Society 2002) for use in clinical trials of various cardiac and pulmonary conditions.
In recent years the 6MWT has been adapted to evaluate functional capacity in neuromuscular diseases and has served as the basis for regulatory approval of a number of drugs for rare diseases, with mean changes in the 6MWT ranging from 28 to 44 meters (Rubin 2002, Wraith 2004, Muenzer 2006).
Additionally, published data from longitudinal natural history studies assessing dystrophinopathy, a disease continuum comprised of DMD and Becker muscular dystrophy, support the utility of the 6MWT as a clinically meaningful endpoint (McDonald C, et al, Muscle & Nerve, December 2010) in DMD.
These data show that boys with DMD experience a significant decline in walking ability compared to healthy boys over one year, suggesting that slowing the loss of walking ability is a major treatment goal.
About the Statistical Methodology
The Mixed Model Repeated Measures (MMRM) test was used for all statistical analyses of the 6MWT results, including for all subgroups. Analysis of Covariance (ANCOVA) for ranked data was used when the assumptions of normality of the dependent variable (the change in 6MWT distance from baseline) were violated.
The inclusion of the two patients with extreme scores due to rapid progression in the ITT population (n=12) resulted in a violation of the normality assumptions of the Change from Baseline in 6MWT data, and thus required the use of ANCOVA for ranked data.
The exclusion of these two patients from the mITT population (n=10) resulted in the 6MWT data becoming normally distributed and the MMRM statistics exhibiting much improved residuals and fit statistics as compared to the ITT population.
As such, the estimated mean values and their associated p-values for the mITT population were slightly different from those for the ITT population.
Sources:
SAREPTA
Action Duchenne
Other resources:
SAREPTA will hold a conference call and broadcast a slide show today at 8:00 a.m. EDT (5:00 a.m. PDT) to discuss these results. The audio conference call may be accessed by dialing 866.356.3093 for domestic callers and 617.597.5381 for international callers. The passcode for the call is 93880948. Please specify to the operator that you would like to join the "Sarepta Therapeutics 48-Week Results Call." To view the slide show while using the audio dial-in please go to the events section of Sarepta's website at www.sareptatherapeutics.com. The call and slide show will also be webcast live under the events section and will be archived there following the call for 90 days. Please connect to Sarepta's website several minutes prior to the start of the broadcast to ensure adequate time for any software download that may be necessary. An audio replay will be available through October 10, 2012 by calling 888.286.8010 or 617.801.6888 and entering access code 67898748.
Read More