In a paper just published online in the journal The Lancet, Craig McDonald and colleagues at 53 study sites in 18 countries describe the clinical benefit of using the drug ataluren for a certain group of patients carrying a specific "nonsense mutation" for Duchenne muscular dystrophy.
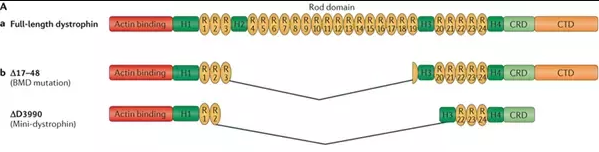
Duchenne muscular dystrophy (DMD) is a progressive and life-limiting X-linked recessive disorder caused by mutations in the DMD gene that result in reduced or absent dystrophin production. Dystrophin is part of the dystrophin–glycoprotein complex, which acts as a scaffold between the actin cytoskeleton and the extracellular matrix and, as such, maintains muscle fibre integrity.
A single nucleotide change in the DNA sequence that introduces a premature stop codon is known as a nonsense mutation, a subset of a major class of premature termination codon (PTC) mutations. Nonsense mutations cause premature termination of translation resulting in the production of truncated polypeptides, which in turn halts the ribosomal translation process at an earlier site than normal, producing a truncated, non-functional protein. Ataluren makes ribosomes less sensitive to premature stop codons (referred to as "read-through"). Ataluren allows for those premature stop codons to be read through and complete the protein:
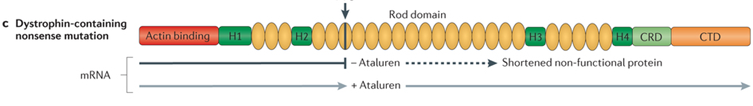
Ataluren has been marketed under the trade name Translarna by PTC Therapeutics (South Plainfield, NJ, USA). The drug allows ribosomal readthrough of premature stop codons, thus enabling production of functional dystrophin that might ameliorate disease progression.
The study, a phase 3, multicentre, randomised, double-blind, placebo-controlled trial (ACT DMD) was sponsored by the New Jersey-based company that developed ataluren, PTC Therapeutics. It included 230 patients, half of whom received the drug therapy over a course of 48 weeks, and the other half who received a placebo.
It assessed the ability of ataluren to stabilise ambulation, with a focus on a prespecified subgroup of patients with ambulatory decline. The primary endpoint of change in 6-min walk distance (6MWD) from baseline to week 48, with a hypothesis of a difference of at least 30 m between ataluren-treated and placebo-treated patients, did not differ significantly between groups in the intention-to-treat population.
Duchenne study participants with the nonsense mutation who had a baseline six-minute walk of between 300 meters and under 400 meters were the subgroup of patients who were the most likely to see the clinical benefits of ataluren.
ACT DMD and the use of ataluren provide a glimmer of hope for the approximately 10–15% of patients with DMD have an underlying nonsense mutation in the DMD gene 2 where the drug seems to prompt a slowing or stabilizing of the disease progression and motor function.
In 2016, the European Medicines Agency gave conditional approval for the drug to be used and, once data demonstrating its ability to stabilise ambulation were obtained, the National Institute for Health and Care Excellence (NICE) agreed reimbursement within a managed access agreement for the treatment of ambulant patients with DMD aged 5 years or older.
Other therapeutic approaches are currently being trialled in patients with DMD, several of which are mutation specific. These approaches include the use of compounds facilitating the upregulation of dystrophin analogues (NCT02858362); exon-skipping techniques with antisense oligonucleotides to convert an out-of-frame mutation into an in-frame mutation, thus allowing partial dystrophin expression; and the use of selective steroid receptor modulators (NCT02760264).
Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial, Craig M McDonald et al., The Lancet, doi: 10.1016/S0140-6736(17)31611-2, published 17 July 2017.
Wood CL, Cheetham T (2017) Treatment of Duchenne muscular dystrophy: first small steps.Lancet():. DOI: 10.1016/S0140-6736(17)31669-0 PMID: 28728957.
Read More
Add comment