-
New treatment of Fukuyama congenital muscular dystrophy
Scientists rescue Fukuyama congenital muscular dystrophy-related brain defects in a new organoid model developed using stem cells from affected patients
Fukuyama congenital muscular dystrophy (FCMD) is a severe genetic neuromuscular disorder affecting the eyes, brain, and muscles. FCMD is caused by mutations disrupting the glycosylation (addition of sugars) of α-dystroglycan (αDG)—a protein required for maintaining muscle integrity. During fetal development, the lack of glycosylation destroys αDG function, leading to severe muscular dystrophy, intellectual disabilities, seizures, and insomnia. Infants born with FCMD have low life expectancies, and patients typically do not survive into their 20s. Despite this, no cure for FCMD has been discovered yet, partly owing to the unavailability of appropriate animal models for research.
In a recent breakthrough, scientists from Japan and the USA have developed the first human disease model of FCMD using stem cells from a patient with FCMD. Their study, published in the peer-reviewed journal iScience, describes the effectiveness of a small compound called Mannan-007 (Mn007) in reducing FCMD-related defects in this model.
For Dr. Mariko Taniguchi-Ikeda, Assistant Professor at the Fujita Health University Hospital and also the lead investigator on this study, finding a cure for FCMD has been a life-long mission. She says, “Having worked with patients with FCMD for more than 20 years, I understood the importance of attempting to cure this disease. But first, we needed a better model to study FCMD in the laboratory.”
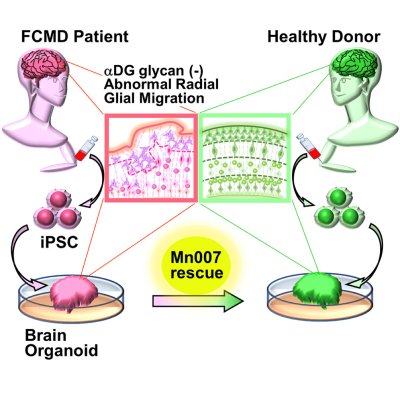
To solve this problem, Dr. Taniguchi-Ikeda’s team transformed pluripotent stem cells derived from an FCMD patient into skeletal muscle and “brain organoids”—artificially grown miniature organs resembling the brain. “Using a brain organoid model derived from pluripotent stem cells, we showed the restoration of defects in the radial architecture of neurons and cortical plate organization of Fukuyama muscular dystrophy,” explains Prof. Takashi Aoi from Kobe University, who collaborated with Dr. Taniguchi-Ikeda on this study.
The brain organoids successfully mimicked the features of FCMD, including the low levels of αDG glycosylation. They also showed impairments in neuronal migration—a process crucial for normal brain development—owing to a disorganized arrangement of guiding cells called radial glia. This new fetal human brain model of FCMD was therefore superior to animal models, which do not fully reproduce FCMD-like brain anomalies.
Encouraged by the successful development of this model, the team set out to test potential treatment options for FCMD. In particular, they investigated whether the small compound Mn007 could reduce the FCMD-like defects observed in the organoids. Dr. Taniguchi-Ikeda explains, “Because of gene mutations, the multi-step process of αDG glycosylation is impaired in FCMD. We hypothesized that the addition of a molecule like Mn007 that can promote αDG glycosylation would rescue the defects observed in FCMD.”
To their delight, their hypothesis was validated. They observed that Mn007 could restore the levels of αDG in both the muscle and brain models tested. Further, brain organoids treated with Mn007 showed partial improvements in radial glial architecture and neuronal migration, resulting in fewer development defects. Their findings therefore highlighted the importance of αDG glycosylation in regulating these processes and demonstrated the potential of Mn007 and similar molecules in therapies for FCMD and related diseases.
This study from Dr. Taniguchi-Ikeda and her team brings a ray of hope for thousands of potential parents worldwide, and especially in Asia, where most cases of FCMD are encountered. “Mn007 can cross over from the blood into the brain. Therefore, it could be a viable prenatal option for restoring normal neuronal migration in patients with FCMD and other diseases caused by glycosylation defects in αDG and reducing their brain-related symptoms. This is an exciting opportunity for clinical translation down the road,” comments Prof. Bennett G. Novitch from the David Geffen School of Medicine at the University of California, who also contributed to this study. His co-author, Dr. Momoko Watanabe from the University of California, Irvine, adds that their findings also offer a valuable opportunity for delineating the molecular mechanisms that lead to migration defects caused by the lack of αDG glycosylation in FCMD.
Overall, the findings of this study are an important landmark in the quest to tackle diseases like FCMD. And while a cure could still be years away, thanks to Dr. Taniguchi-Ikeda and her team, the future of therapies targeting FCMD and other related diseases now looks brighter than ever.
Source: News Release
DOI: https://doi.org/10.1016/j.isci.2021.103140
Upcoming Events
Join Our Newsletter
Why Join the ICNA?