Comparitive genomic hybridization (CGH) or Chromosomal microarray analysis(CMA) is a modern cytogenetic technique for analyzing variations in DNA copy-number. Alteration in DNA copy number is one of the many ways in which modification of gene expression and function occurs. The technology was initially used for studying chromosomal imbalances in solid tumours.
 Large-scale copy number variations in the human genome leading to chromosomal imbalances comprise approximately 12% of the entire genome and some 10% of all known genes [Moechsler 2008].A copy number variation (CNV)is defined as a DNA segment of longer than 1 kb with a variable copy number compared with a normal reference genome. Copy number variations are bounded by a stretch of similar DNA sequences called low-copy repeats or segmental duplications (or interspersed duplication blocks) which may potentially act as recombination hot spots.It should be noted that variations in DNA copy number though associated with disease states, can also be seen in healthy individuals
Large-scale copy number variations in the human genome leading to chromosomal imbalances comprise approximately 12% of the entire genome and some 10% of all known genes [Moechsler 2008].A copy number variation (CNV)is defined as a DNA segment of longer than 1 kb with a variable copy number compared with a normal reference genome. Copy number variations are bounded by a stretch of similar DNA sequences called low-copy repeats or segmental duplications (or interspersed duplication blocks) which may potentially act as recombination hot spots.It should be noted that variations in DNA copy number though associated with disease states, can also be seen in healthy individuals
In recent years chromosomal microarray analysis have come into routine use in child neurology to enable genome-wide evaluation in developmental delay, mental retardation, and autistic spectrum disorders. Chromosomal microarray results should be interpreted with care with the help of published genetic databases,because of the relatively large number of benign CNV in normal individuals. Early consultation should be made with a clinical geneticist.
Microarray technology
- Conventional karyotyping detects chromosomal abnormalities of 5 Mb and larger, including balanced translocations. Even high-resolution chromosome analysis (700-850 bands) would not detect an aberration smaller than 3 Mb.While the diagnostic yield of conventional karyotyping in developmental delay or mental retardation is in the order of 3.7-9.5% by chromosomal microarray analysis the yield has been reported to increase upto 15-20%.
- The DNA samples from a patient and a reference genome are labeled with different fluorescent dyes and are cohybridized to a known genomic sequence. Differences in relative fluorescence intensities of hybridized DNA on the microarray reflect differences in copy number between the genomes of the patient and the reference.
- Bacterial artificial chromosomes (150-200 kb in size) has a higher detection rate than conventional karyotyping, but is unreliable in detecting abnormalities smaller than the bacterial chromosome itself.The coverage of the bacterial artificial chromosome array over the genome is also limited [Bejjani 2005] [Veltman 2006]
- Microarrays based on oligonucleotide probes of various sizes have been developed which can detect variations in copy number of genomic sequence as small as 99 kb [Sebat 2007]
- More advance single-nucleotide polymorphism arrays bypass the need for a labeled reference genome during hybridization and instead compares fluorescence intensities of up to a million loci with those obtained from a series of normal control genomes. Single-nucleotide polymorphism arrays have successfully detected copy number variations (CNVs) of DNA sequences as small as 178 kb [Friedman 2006]. Thus single-nucleotide polymorphisms help in linkage analysis studies adn also allow detection of copy-neutral loss of heterozygosity correlated with uniparental disomy [Altug-Teber 2005] and consanguinity [DeLucca 1991]
- Combining the information of CNV and the minor-allele frequency at the corresponding loci, one can also detect mosaicism in DNA copy number changes, to levels of mosaicism as low as 10-20%.
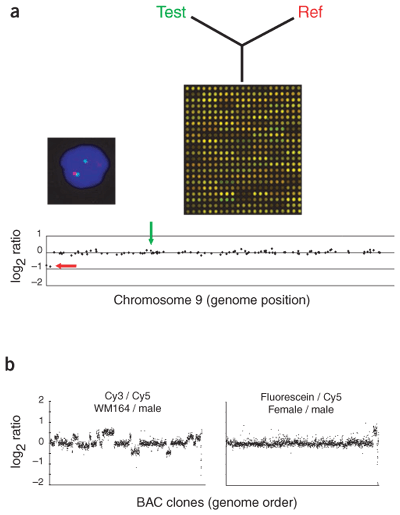
Overview of array CGH.
(a) Genomic DNA from two cell populations is differentially labeled and hybridized to a microarray. The fluorescent ratios on each array spot are calculated and normalized so that the median log2 ratio is 0.
Plotting of the data for chromosome 9 from pter to qter shows that most elements have a ratio near 0. The two elements nearest pter have a ratio near -1, indicating a reduction by a factor of two in copy number. FISH with a red-labeled probe for the deleted region and a green-labeled control probe (genome locations indicated by the red and green arrows on the ratio profile) shows that the cells contain two copies bound by the green probe and only one bound by the red, consistent with the array CGH analysis7.
(b) Simultaneous comparison of three genomes. Cy3-labeled genomic DNA from melanoma cell line WM-164, Cy5-labeled normal male genomic DNA and fluorescein-labeled normal female genomic DNA were simultaneously hybridized to a BAC array and imaged using a custom-build CCD imaging system (D.P., D.G.A. et al., unpublished data).
The left panel shows the Cy3/Cy5 ratio (cell line/normal male) for each clone plotted in genome order. A large number of aberrations are evident in this nondiploid cell line. The right panel shows the fluorescein/Cy5 ratio (normal female/normal male) with the X chromosome copy-number difference evident. Multigenome hybridizations of this type permit more efficient use of arrays and the inclusion of a control with each unknown specimen.
Unpublished data courtesy of A. Estep (University of California San Francisco, California, USA). source:Nature Genetics 37, S11 - S17 (2005)
Genetic evaluation of neurodevelopmental disability
The American Academy of Neurology, the American Academy of Pediatrics, and the American College of Medical Genetics have established guidelines for the evaluation of children with developmental delay and mental retardation that include recommendations for genetic testing.
| Current recommendations for the genetic evaluation of neurodevelopmental disability | ||
|---|---|---|
| Professional Society | Recommendation | Reference |
| American Academy of Neurology; Child Neurology Society | Karyotyping; molecular fragile X testing; selected subtelomeric FISH testing; Rett Syndrome testing where appropriate | Shevell et al., 2003 |
| American College of Medical Genetics | Karyotyping (>550 bands); FISH testing for specific syndromes; consider subtelomeric FISH testing | Shaffer, 2005 |
| American Academy of Pediatrics | Karyotyping; molecular fragile X testing; subtelomeric FISH testing; focused metabolic testing; focused single-gene testing | Moeschler et al., 2006 |
Several microdeletions and microduplications have been recently described in children with unexplained mental retardation, autism, or congenital anomalies and in unaffected persons. These imbalances remain undetectable using routine karyotype analysis.The use of array comparative genomic hybridization is thus replacing the use of fluorescent in-situ hybridization techniques for the child with idiopathic global developmental delay or intellectual disability.
Chromosomal Microarray testing
The interpretation of microarray data is complicated by the fact that copy number variations frequently involving genes are also seen in healthy individuals. Wong et al 2007 using a whole-genome array comparative genomic hybridization assay identified 3,654 autosomal segmental CNVs, 800 of which appeared at a frequency of at least 3%. These copy number variations may account for the phenotypic diversity of the human genome.
Known Copy Number Variations (CNV) are now maintained in the public domain on online databases (http://www.projects.ca/variation/ andhttp://sanger.ac.uk/humgen/cnv/). Currently there are no standards for either naming or describing these CNVs.
Algorithm for Chromosomal Microarray Interpretation
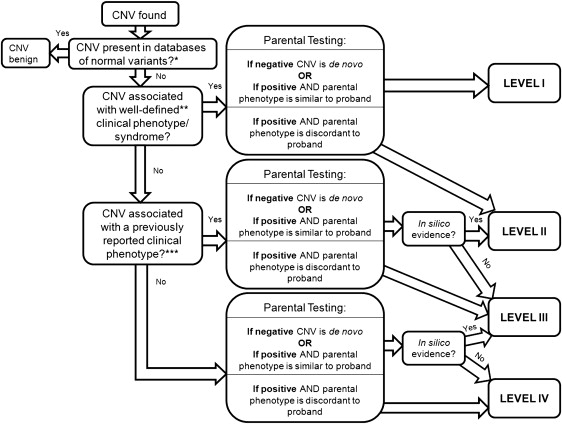
- Whenever a CNV is identified in a proband, testing of both parents is necessary to distinguish whether the CNV is de novo or inherited. This is typically performed using FISH techniques, and is commonly offered at no additional cost by commercial laboratories that perform chromosomal microarray testing. Nonpaternity or adoption can render the inheritance pattern unknown. An apparently de novo imbalance may be due to a balanced rearrangement from a parent.
- If inherited, careful assessment of the parental phenotype is important as a basis from which to infer whether the CNV is correlated with the phenotype. This can be further complicated by incomplete penetrance and variable expressivity.
- Inorder to establish that a CNV is responsbile for the patient's phenotype, the same or overlapping CNV should be seen in patients affected with similar phenotypes but should be absent from a control population. Variable penetrance and expressivity may again make this difficult
- Modifier genes can also diversify the spectrum of phenotypes associated with a genotypic anomaly. Copy number variations in genomic regions that are gene rich are more likely to be clinically significant, whereas CNV in a gene-poor region is less likely to be pathogenic.
- Obvious bias in parental origin of an inherited CNV may suggest an epigenetic mechanism of a disease.
Scenario 1:A CNV is found that is associated with a well-defined clinical phenotype or syndrome. This would provide level I evidence that the chromosomal microarray result is related to the patient's phenotype, and often there is a considerable quantity of published natural history data to provide to the family. Parental samples should still be obtained (to confirm that the CNV is not carried with incomplete penetrance by a parent), because this may be relevant for future offspring. The parental karyotype should also be examined, to rule out a parental balanced translocation at the locus in question and a clinical geneticist referral made for futher counselling.
Scenario 2: A CNV is found in the proband and is also found in one parent with either a normal phenotype or some other phenotype
In this case, an analysis of the genes contained in the CNV should be performed using in silico techniques such as the University of California at Santa Cruz Genome Browser (http://www.genome.ucsc.edu/) coupled with a search of PubMed (http://www.ncbi.nlm.nih.gov/pubmed/), and DECIPHER (https://decipher.sanger.ac.uk/), to see whether abnormalities in those genes have been associated with phenotype similar to the proband. If there is available evidence to suggest that the CNV is associated in some way with the patient's phenotype, variable penetrance or other modifying genes may be active in the pedigree, and only level II evidence can be cited for a genotype-phenotype association.
If the parent identified with the CNV in question, is not normal but has a phenotype different from the proband then Level II evidence allows for the variation in genotype-phenotype association in this scenario. The one gene-one disease model (or one rearrangement, one disease) has been challenged recently, based on the association of 1q21.1 rearrangement with a broad spectrum of phenotypes, and the genotype-first clinical diagnostic approach has been proposed [1] and [3].
Scenario 3: A CNV is found that has not been previously described, and whose clinical phenotype is unclear.
An in silico analysis of the CNV using the University of California at Santa Cruz Genome Browser should be performed, as well as a search of PubMed, to see whether abnormalities in those genes have been associated with phenotype similar to the proband.Additional individualized testing, such as quantitative real-time polymerase chain reaction or multiple ligation-dependent polymorphism analysis, to assess copy number on an exon-by-exon basis within a candidate gene to confirm that the CNV is in fact associated with the proband's presentation.
The CNV data and associated clinical findings should be submitted to online databases such as DECIPHER to help a body of evidence concerning any given CNV (https://decipher.sanger.ac.uk/). Until more evidence is accumulated in the literature, only level III evidence can be provided for a genotype-phenotype association in such situations
If a reciprocal microduplication is ascertained, but only the accompanying microdeletion is described in the literature, again caution should be taken and multiple cases of the CNV in question should be ascertained before conclusions can be drawn about the clinical phenotype or phenotypes. Reciprocal CNV phenotypes can differ widely from one another, as has been shown in studies of the syndromes associated with 22q11.2 microduplication and microdeletion.
Scenario 4: Multiple CNVs are found in the proband, but not in either parent
If one CNV found is associated with a literature-described phenotype, but one or more other CNVs are not counseling can be directed by the literature associated with the known level I genotype-phenotype correlation. If none of the CNVs uncovered are described in the literature, another in silico search is indicated using the University of California at Santa Cruz Genome Browser to identify genes located in the CNVs, followed by a search of PubMed, for published phenotypic data associated with abnormalities in those genes. Until individuals with gene mutations that recaptitulate features of the syndrome associated with a CNV, only level III evidence can be cited regarding genotype-phenotype correlation.
Scenario 5: Multiple CNVs are found in the proband, with one or more also found in either normal parent
An in silico analysis of the candidate genes within the CNV may help to clarify matters, by identifying genes in a CNV region not shared by the parent with an associated literature strongly recapitulating the patient's phenotype.If this is not possible the chromosomal microarray results would be uninterpretable (level IV evidence of genotype-phenotype correlation) . Individual clinical judgment should be used for genetic counselling.
Prenatal genetic counseling for CNV will remain difficult until more, genotype-phenotype correlation data become available through ongoing research.
References
- Moeschler JB (March 2008). "Genetic evaluation of intellectual disabilities". Semin Pediatr Neurol 15 (1): 2–9. doi:10.1016/j.spen.2008.01.002. PMID 18342255.
- Pinkel D, Albertson DG (June 2005). "Array comparative genomic hybridization and its applications in cancer". Nat. Genet. 37 Suppl: S11–7. doi:10.1038/ng1569. PMID15920524.
- Bejjani BA, Saleki R, Ballif BC, Rorem EA, Sundin K, Theisen A, Kashork CD, Shaffer LG (April 2005). "Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more?". Am. J. Med. Genet. A 134 (3): 259–67. doi:10.1002/ajmg.a.30621. PMID15723295.
- Veltman JA, de Vries BB (November 2006). "Diagnostic genome profiling: unbiased whole genome or targeted analysis?". J Mol Diagn 8 (5): 534–7; discussion 537–9. PMID17065419. PMC1876178.
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimäki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M (April 2007). "Strong association of de novo copy number mutations with autism". [[Science (journal)|Science]] 316 (5823): 445–9. doi:10.1126/science.1138659. PMID17363630.
- Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Armstrong L, Asano J, Bailey DK, Barber S, Birch P, Brown-John M, Cao M, Chan S, Charest DL, Farnoud N, Fernandes N, Flibotte S, Go A, Gibson WT, Holt RA, Jones SJ, Kennedy GC, Krzywinski M, Langlois S, Li HI, McGillivray BC, Nayar T, Pugh TJ, Rajcan-Separovic E, Schein JE, Schnerch A, Siddiqui A, Van Allen MI, Wilson G, Yong SL, Zahir F, Eydoux P, Marra MA (September 2006). "Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation". Am. J. Hum. Genet. 79 (3): 500–13. doi:10.1086/507471. PMID16909388. PMC1559542.
- Altug-Teber O, Dufke A, Poths S, Mau-Holzmann UA, Bastepe M, Colleaux L, Cormier-Daire V, Eggermann T, Gillessen-Kaesbach G, Bonin M, Riess O (August 2005). "A rapid microarray based whole genome analysis for detection of uniparental disomy". Hum. Mutat. 26 (2): 153–9.doi:10.1002/humu.20198. PMID15968682.
- Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, Majnemer A, Noetzel M, Sheth RD (February 2003). "Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society". Neurology 60 (3): 367–80. PMID12578916.
- Shaffer LG (2005). "American College of Medical Genetics guideline on the cytogenetic evaluation of the individual with developmental delay or mental retardation". Genet. Med. 7 (9): 650–4. PMID16301868.
- Moeschler JB, Shevell M (June 2006). "Clinical genetic evaluation of the child with mental retardation or developmental delays". Pediatrics 117 (6): 2304–16. doi:10.1542/peds.2006-1006. PMID16740881.
- Wong KK, deLeeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, MacAulay C, Ng RT, Brown CJ, Eichler EE, Lam WL (January 2007). "A comprehensive analysis of common copy-number variations in the human genome". Am. J. Hum. Genet. 80 (1): 91–104.doi:10.1086/510560. PMID17160897. PMC1785303.
Further Reading
Related
Cite this: ICNApedia contributors.Comparitive genomic hybridization (CGH) . ICNApedia, The Child Neurology Knowledge Environment. 30 June 2024. Available at: https://icnapedia.org/knowledgebase/articles/comparitive-genomic-hybridization-cgh Accessed 30 June 2024.