Lennox-Gastaut Syndrome (LGS) was first characterized by Dr. William G. Lennox as a form of epilepsy known as a "Petit mal variant." In 1966, the Marseille School in France provided a more detailed description of the syndrome. Dr. Henri Gastaut and his colleagues introduced the term "Lennox syndrome" to refer to a type of epilepsy that begins in childhood and is characterized by recurrent tonic and absence seizures. In 1989, the International League Against Epilepsy (ILAE) issued a more precise diagnosis of Lennox-Gastaut Syndrome (LGS). The illness is categorized as an epileptic encephalopathy, which means that the epileptic activity has a role in causing cognitive and behavioral abnormalities.
Epidemiology
The prevalence of Lennox-Gastaut Syndrome (LGS) is estimated to be between 1% and 2% of all epilepsy patients, and between 1% and 10% of childhood epilepsies. This wide range is likely due to variations in diagnostic criteria and research settings across different studies.
Studies on Prevalence
United States Study
A population-based study in the USA defined children as having LGS if they exhibited multiple seizure types before the age of 11, with at least one type resulting in falls, and an EEG showing slow spike-wave complexes (< 2.5 Hz). Intellectual disability (ID) was not used as a diagnostic criterion. The lifetime prevalence of LGS at age 10 was found to be 0.26 per 1,000 children. In this study, 91% of those with LGS had an ID, and 17% of all children with profound ID (IQ < 20) had LGS. LGS accounted for approximately 4% of all childhood epilepsies (Trevathan E et al., 1997).
Iranian Study
In a study from Iran, 5.4% of all epilepsy patients (both children and adults) were diagnosed with LGS. Given that LGS rarely remits, prevalence studies tend to report higher frequencies than incidence studies (Camfield PR, 2011)
Incidence Studies
In a population-based study of children with new-onset epilepsy, 12% had symptomatic generalized epilepsies, with only 4% of these cases being LGS. The incidence of LGS among all new-onset epilepsies was 0.6% (Camfield CS et al., 1996).
Age of Onset
The onset of LGS usually occurs before the age of 8 years. However, late-onset LGS has been documented in about 10% of patients, with the syndrome starting after 8 years of age in one study, and 16% in another.
Gender Distribution
Males are more frequently affected by LGS than females. One study reported a male-to-female ratio of 1.6, while another reported a ratio of 1.49. The reason for this male predominance is currently unclear.
Etiology of Lennox-Gastaut Syndrome (LGS)
Identifiable Causes
The etiology of Lennox-Gastaut Syndrome (LGS) is often categorized into two groups: identifiable (genetic-structural-metabolic) and unknown causes. Approximately 65 to 75% of LGS patients have an identifiable cause. These causes may include:
- Brain damage (e.g., birth asphyxia, head injuries)
- Tuberous sclerosis complex
- Congenital central nervous system (CNS) infections
- Brain malformations
- Hereditary metabolic disorders
A previous study reported epilepsy risk factors as follows:
- Perinatal complications (25%), including hypoxic-ischemic insults, sepsis, low birth weight, and hyperbilirubinemia
- CNS infections (3.7%)
- Significant head trauma (less than 1%)
Identifiable causes are typically due to a static brain disorder, with progressive or metabolic disorders being rare. Brain magnetic resonance imaging (MRI) is a crucial diagnostic tool for identifying the etiology of LGS. Modern 3-T MRI scanners and sequences can detect subtle abnormalities in patients with LGS, even when previous brain MRI scans were unremarkable or inconclusive.
Unknown Causes
The group of LGS patients with unknown causes accounts for approximately 25 to 35% of cases. The attribution of "unknown" depends significantly on the sophistication of the investigations. In one study, 70% of adult patients had LGS of unknown cause (cryptogenic) (Ferlazzo E etal., 2010). Another study characterized LGS of unknown cause by phenotypic analysis of patients and their parents, enrolling 135 patients from 19 centers in the USA and Australia (Widdess-Walsh P et al., 2013). The study concluded that LGS of unknown cause has distinctive characteristics, including a broad age range of onset, male predominance, and often normal development before the onset of seizures.
When LGS has no apparent cause, a genetic predisposition or etiology is likely. Several genetic factors have been reported in association with LGS, including:
- Copy number variants
- SCN1A mutations
- CHD2 mutations
- De novo missense mutation in the forkhead box G1 (FOXG1) gene
- Mutations in dynamin 1 (DNM1), encoding the presynaptic protein DNM1
A study highlighted the genetic heterogeneity of LGS and introduced rare copy number variants as significant risk factors (Lund C etal., 2013). The CHD2 gene, located on 15q26.1, is associated with various human developmental disorders and is likely important in the etiological spectrum of LGS.
Implications of Genetic Studies
Epileptic encephalopathies share overlapping features and may evolve from one to another. Identifying genetic etiology can aid clinicians in predicting the prognosis of patients with LGS. For example, LGS may evolve from West Syndrome/infantile spasms in about 20% of patients. Results from genetic studies may have significant implications for therapeutic choices, prognosis, and genetic counseling for affected children and their families
Secondary Network Epilepsy
Lennox-Gastaut Syndrome (LGS) is often considered a secondary network epilepsy. The typical epileptic manifestations, including tonic seizures, slow spike-wave (SSW) complexes, and generalized paroxysmal fast activity (GPFA), reflect network dysfunction rather than a specific initiating process, such as a focal lesion (Archer JS etal., 2014).
fMRI and EEG Studies
A study recorded GPFA and SSW complexes simultaneously with functional MRI (fMRI) in patients with LGS. The results showed that generalized paroxysmal fast activity events exhibited almost uniform increases in blood oxygen level-dependent (BOLD) signal in association cortical areas, brainstem, basal ganglia, and thalamus. In contrast, SSW complexes demonstrated a different pattern of BOLD signal change, with many areas of decreased BOLD signal primarily in primary cortical areas. The study concluded that GPFA is associated with activity in a diffuse network, including association cortices and subcortical structures, whereas SSW complexes involve distinct patterns of cortical and subcortical activations and deactivations (Pillay N etal., 2013).
Cognitive Network Interactions
Another EEG-fMRI study of LGS patients examined cognitive networks and found reduced within-network integration, including weaker connectivity within the default mode network. The study also observed impaired between-network segregation, with stronger connectivity between the default mode and dorsal attention networks (Warren AE, 2016). Abnormal interactions were noted during fMRI periods both with and without epileptiform discharges on scalp EEG. The authors concluded that cognitive network interactions are persistently abnormal in patients with LGS. These findings suggest that the epileptic process in LGS may initiate and sustain abnormal network behavior.
Clinical Characteristics of Lennox-Gastaut Syndrome (LGS)
Intellectual and Psychosocial Dysfunction
Lennox-Gastaut Syndrome (LGS) significantly impacts the intellectual function of affected patients. Cognitive impairments are clinically apparent in many patients (20 to 60%) at the time of diagnosis. These impairments typically become more pronounced over time, with serious intellectual problems noted in most patients (75 to 99%) within five years of onset. A minority (10–20%) of affected children remain within the accepted limits of normal intellectual function but often face difficulties in everyday life due to a slowing of mental processing. Favorable cognitive outcomes are more likely in patients with a later age of onset.
Alongside cognitive problems, many LGS patients experience behavioral and psychiatric issues. Attention problems, aggression, and autistic behaviors are common and present significant challenges for families and caregivers. These behavioral problems may arise from a combination of factors, including the epilepsy itself, abnormal network characteristics, and the effects of medications.
Seizure Types
Tonic Seizures:
- The most characteristic type of seizure in LGS.
- Considered a prerequisite for diagnosis by some experts.
- Occurrence varies among studies.
- Often underestimated due to frequent occurrence during non-rapid eye movement (non-REM) sleep.
- Higher incidence found in studies with EEG recordings during sleep.
- Periictal single-photon emission computed tomography (SPECT) studies suggest tonic seizures result from activity in a network involving bilateral frontal and parietal association areas and the pons.
Atypical Absences:
- Second most common type of seizure in LGS.
- Difficult to recognize and accurately estimate their frequency.
- Characterized by gradual onset and termination.
- Diminished cognitive abilities in patients may limit responsiveness.
- Reliable diagnosis requires video-EEG monitoring.
Epileptic Drop Attacks:
- Particularly hazardous, occurring in more than half of LGS patients.
- Can result from tonic, atonic, or myoclonic seizures.
- Also observed in other epilepsy syndromes, making their presence non-diagnostic for LGS.
Nonconvulsive Status Epilepticus (NCSE):
- Affects about two-thirds of LGS patients.
- Consists of prolonged atypical absences with varying degrees of altered consciousness, periodically interrupted by brief tonic seizures.
- Can last from hours to weeks.
- Difficult to recognize in patients with severe cognitive impairment.
- Suspected to be a major contributor to intellectual impairment.
- Identified risk factors for severe intellectual disability include NCSE, previous diagnosis of West Syndrome, symptomatic etiology of epilepsy, and early age of epilepsy onset.
Other Seizure Types:
- Myoclonic seizures, focal seizures, generalized tonic-clonic seizures, and unilateral clonic seizures are also common.
- These seizures typically occur in the later stages of LGS but may sometimes precede the core seizures, complicating diagnosis.
These findings highlight the complex and multifaceted nature of LGS, necessitating comprehensive and multidisciplinary management to address the diverse challenges faced by patients and their caregivers.
Electroencephalographic Features
The EEG background activity in Lennox-Gastaut Syndrome (LGS) is typically abnormal, characterized by a diffuse increase in slow waves (theta and delta) and a slow or absent posterior dominant rhythm. Key EEG features include:
Slow Spike-and-Wave (SSW) Complexes
- Frequency: Less than 2.5 Hz
- Background Activity: Abnormally slow
- Characteristics: Not every slow wave is preceded by a spike or sharp wave, and bursts may be irregular without clear onset and offset. Bursts of SSW complexes may "come and go," making the distinction between ictal and interictal discharges difficult.
- Associated Seizures: Clinically apparent atypical absence seizures always have an associated SSW burst.
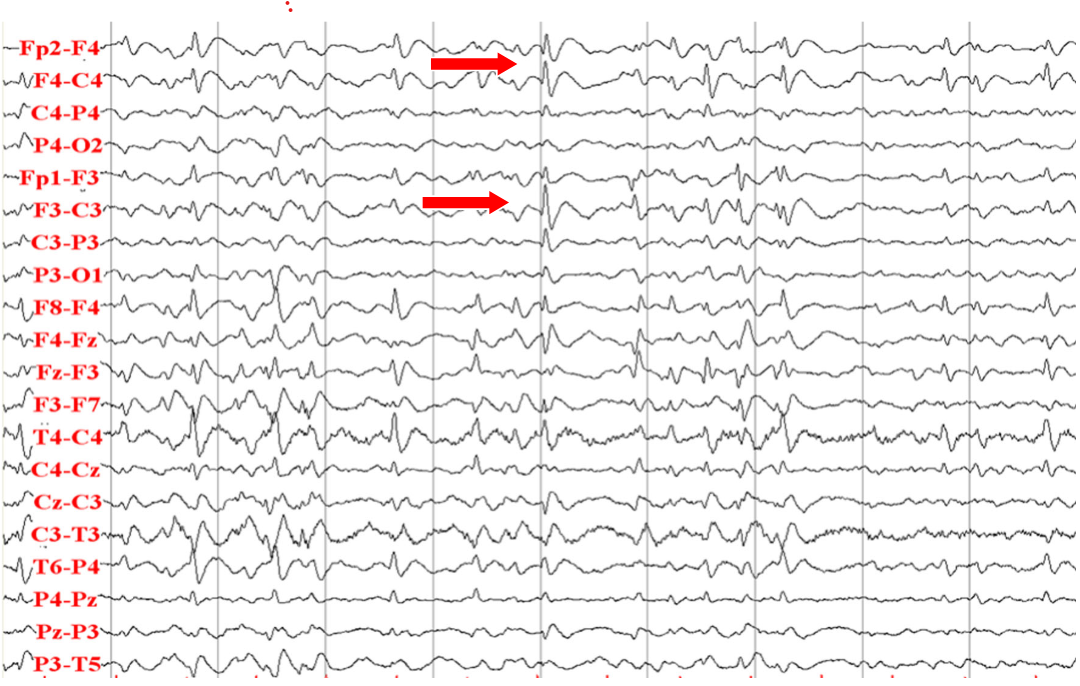 Slow spike-waves (arrows) and diffusely slow background in Lennox-Gastaut syndrome (source: Asaadi Pooya AA et al., 2017)
Slow spike-waves (arrows) and diffusely slow background in Lennox-Gastaut syndrome (source: Asaadi Pooya AA et al., 2017)
A study involving simultaneous fMRI and EEG recordings in nine LGS patients concluded that SSW complexes differ from typical generalized spike-and-wave complexes seen in genetic generalized epilepsies. Differences include deactivation in primary cortical areas, variability of the pattern, inconsistency of thalamic activation, and occasional positive activation in the caudate and basal ganglia. However, not all LGS patients exhibit SSW complexes in their EEGs, with about 13% of patients lacking SSWs (Pillay N et al., 2013).
Generalized Paroxysmal Fast Activity (GPFA)
- Frequency: 10–25 Hz
- Occurrence: Usually recorded during slow-wave sleep.
- Characteristics: Bursts of diffuse or bilateral fast rhythm patterns lasting a few seconds and recurring at brief intervals. These bursts are similar to those seen in tonic seizures with a recruiting rhythm (initial lowering of amplitude followed by a gradual increase in amplitude).
In a study recording GPFA with fMRI in six LGS patients, generalized paroxysmal fast activity showed activation across broad areas of the cortex, sparing primary cortices. GPFA also showed increased BOLD signal in several subcortical structures, including the thalamus, basal ganglia, and brainstem, all of which have broad connections to the association cortices (Pillay N, et al., 2013).
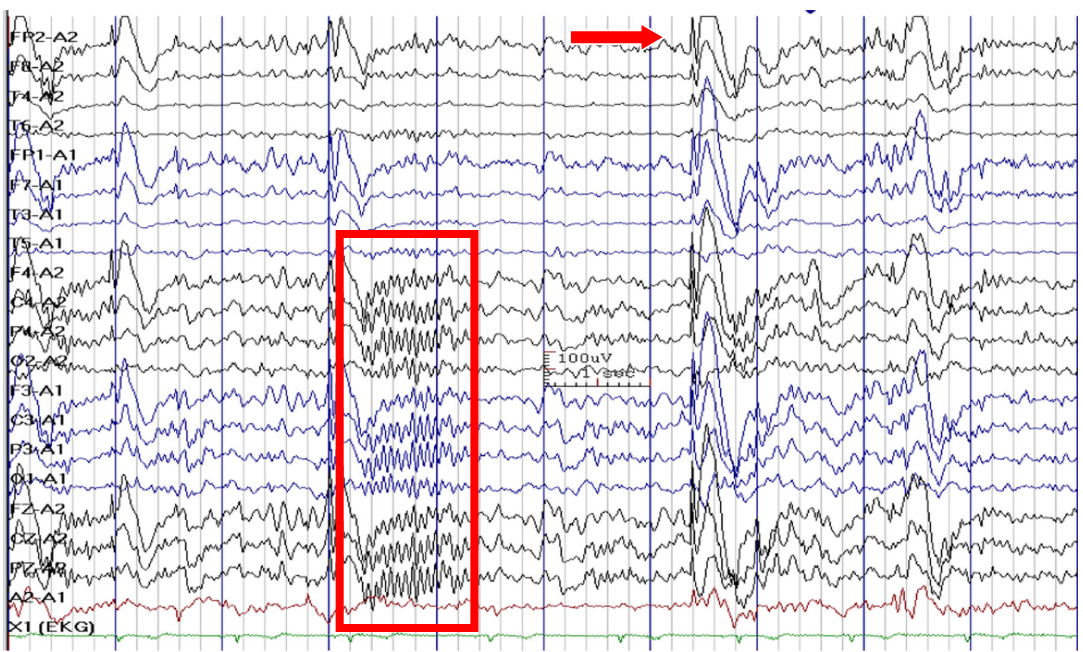 Generalized paroxysmal fast activity (box) and slow spike-waves (arrow) in Lennox-Gastaut syndrome. (Asadi-Pooya AA et al., 2017)
Generalized paroxysmal fast activity (box) and slow spike-waves (arrow) in Lennox-Gastaut syndrome. (Asadi-Pooya AA et al., 2017)
LGS is characterized by abnormal EEG features, including slow spike-and-wave complexes and generalized paroxysmal fast activity. These features reflect the complex network dysfunction underlying the syndrome and are essential for diagnosing and understanding the pathophysiology of LGS.
Differential Diagnosis
The diagnosis of Lennox-Gastaut Syndrome (LGS) is based on a combination of electroclinical criteria, including the predominance of tonic seizures and specific EEG patterns. However, these features may not be present at the onset of the syndrome. Moreover, several other epilepsy syndromes can share one or more criteria with LGS, complicating the differential diagnosis.
Syndromes with Overlapping Features
- Dravet Syndrome
- Myoclonic-Atonic Epilepsy (Doose Syndrome)
- Atypical Benign Focal Epilepsy of Childhood
- West Syndrome
Because LGS can evolve from other syndromes, particularly West Syndrome, the diagnosis may only become apparent after several years of follow-up. Longitudinal studies indicate that the typical features of LGS during childhood can evolve and change over time. In adulthood, it might be challenging to recognize LGS in a previously undiagnosed patient.
Evolution of LGS Over Time
- Childhood: Typical electroclinical features, including frequent and varied seizure types.
- Adulthood: More than half of the patients no longer exhibit all the clinical and EEG characteristics used for diagnosing LGS. The frequency, severity, and variety of seizure types generally decrease, although tonic seizures often persist. Only a minority of adults have slow spike-and-wave (SSW) complexes in their EEG, but the presence of generalized paroxysmal fast activity (GPFA) during sleep remains relatively consistent.
- Cognitive and Behavioral Impact: By adulthood, over 90% of LGS patients have moderate to severe cognitive impairment, often associated with behavioral problems, significantly impacting their social life.
Diagnostic Approach
The diagnosis of LGS should be based on:
- Detailed Clinical History: Including both past and present medical information.
- Physical Examination
- Electroencephalography (EEG): At a minimum, an awake and sleep EEG is required. Overnight video-EEG monitoring can be particularly helpful in confirming the diagnosis.
Accurate diagnosis of LGS relies on recognizing its distinct electroclinical features and understanding its evolution over time. Due to the overlap with other epilepsy syndromes and the changes in presentation from childhood to adulthood, a thorough and longitudinal approach is essential for correct diagnosis and management.
Treatment of Lennox-Gastaut Syndrome (LGS)
Seizure Management
The seizures in Lennox-Gastaut Syndrome (LGS) are typically resistant and intractable to antiepileptic drugs (AEDs), making complete seizure control and resolution of intellectual and psychosocial dysfunction often unachievable. The primary goal in managing LGS is to reduce the frequency of the most incapacitating and injurious seizures, such as drop attacks and tonic-clonic seizures.
Antiepileptic Drugs
Effective AEDs:
- Clobazam: Effective in decreasing drop attacks; generally well-tolerated with fewer sedative effects compared to other benzodiazepines.
- Felbamate: Effective but with significant risks of adverse effects like fatal aplastic anemia and liver failure.
- Lamotrigine: Effective in reducing tonic-clonic seizures and drop attacks; well-tolerated but may exacerbate myoclonic seizures.
- Rufinamide: Approved for adjunctive treatment in children 4 years and older; effective against multiple seizure types.
- Topiramate: Broad-spectrum AED; associated with weight loss and cognitive slowing.
First-Line Drugs:
- Valproate: Often considered the first choice; effective against myoclonic, atypical absence, and atonic seizures but associated with gastrointestinal upset, weight gain, and rare serious effects like hepatotoxicity and pancreatitis.
- Lamotrigine: Generally well-tolerated with common side effects including skin reactions and drowsiness.
- Topiramate: Effective but associated with cognitive slowing and a higher incidence of renal stones.
Adjunctive and Alternative AEDs:
- Levetiracetam, clobazam, nitrazepam, and zonisamide: Reported to be effective in LGS.
- AEDs to Avoid: Phenytoin, carbamazepine, oxcarbazepine, gabapentin, vigabatrin, and tiagabine can aggravate myoclonus or absence seizures.
Specific AED Guidelines:
- Valproate: Starting dose is 7–10 mg/kg/day, increasing gradually with a maximum dose of 60 mg/kg/day or 3000 mg/day.
- Topiramate: Starting dose varies by age, with gradual increases; maximum dose up to 1600 mg/day in adolescents and adults.
- Lamotrigine: Initiate at low doses, gradually increasing to avoid skin rash; maintenance dose typically 200–400 mg/day.
- Clobazam: Starting dose is 5–10 mg/day, with gradual increases; maintenance dose ranges from 20-40 mg/day.
- Rufinamide: Starting dose 10 mg/kg/day for children, 400-800 mg/day for adults, with gradual increases to a maximum dose of 3200 mg/day.
Alternative Treatments
- Cannabidiol: May reduce seizure frequency; more studies needed.
- Steroids: Corticosteroid therapy may reduce seizures but lacks large randomized trials.
- Intravenous Immunoglobulin (IVIG): May reduce seizures but lacks large randomized trials.
Key Points for Prescribing AEDs
- Personalize Treatment: Choose the specific agent based on the patient's profile, comorbidities, potential adverse effects, and drug interactions.
- Gradual Titration: Start doses gradually to improve tolerability and reduce adverse effects. For urgent seizure control, some AEDs can be rapidly titrated.
- Simple Regimens: Prefer once- or twice-daily dosing to improve adherence.
- Monitor Adverse Effects: Question family members and close friends about cognitive or behavioral side effects.
- Treatment Goals: Aim to prevent the most disabling seizures and avoid adverse effects. Adjust therapy if goals are not met. If the first drug fails, switch to monotherapy with a new agent. If the second drug fails, consider adding a second drug.
Ketogenic Diet in the Treatment of Lennox-Gastaut Syndrome (LGS)
The ketogenic diet is an effective and well-tolerated treatment option for patients with Lennox-Gastaut Syndrome (LGS), including those with structural disease and those with unknown causes. This diet should be considered early in the course of the syndrome.
Efficacy
- In one study, over half of the children on the ketogenic diet showed a greater than 50% reduction in seizures (Caraballo RH et al., 2014).
- 20% of these children achieved seizure freedom.
- Patients who responded well to the diet did not experience further mental deterioration.
- Interictal epileptiform abnormalities improved in most patients who had a seizure reduction of more than 75%.
Adverse Effects
While the ketogenic diet has potential adverse effects, the risk of serious adverse events is low. Common adverse effects include:
- Constipation
- Vomiting
- Abdominal pain
- Lack of energy
- Hunger
- Hypercholesterolemia
- Mineral deficiencies
- Acidosis
- Effects on growth
The rigidity of the ketogenic diet and the difficulties associated with lifestyle changes may pose challenges in maintaining the diet.
Alternative Diets
There is evidence supporting the efficacy and tolerability of other dietary approaches in patients with LGS, including:
- Modified Atkins Diet
- Low Glycemic Index Diet
These diets may provide similar benefits with potentially fewer challenges in implementation and maintenance compared to the traditional ketogenic diet.
Surgical Treatment of Lennox-Gastaut Syndrome (LGS)
Overview
Despite ongoing research into drug treatments for LGS, outcomes with chronic administration of antiepileptic drugs (AEDs) remain disappointing. LGS is generally drug-resistant, leading to poor prognoses. For patients with drug resistance, surgical intervention is a further therapeutic option. Several presurgical investigations are required, including video-EEG with natural sleep recording, magnetic resonance imaging (MRI) with a specific epilepsy protocol, and age-appropriate neuropsychological assessment.
Resective Brain Surgery
- Indication: Resective brain surgery can be successful in controlling seizures where and when seizure foci are removable.
- Considerations: Patients with LGS do not necessarily need all their epileptiform discharges to come from one brain area. Successful surgical outcomes have been reported in patients with predominantly focal MRI abnormalities.
- Localization: Successful resective surgery requires localizing the epileptogenic region using clinical history, video-EEG monitoring, MRI, PET, SPECT, MEG, and electrocorticography, as well as determining its relation to the eloquent cortex using Wada test, functional MRI, and brain stimulation.
- Limitations: Resective brain surgery is rarely an option for LGS patients, who often have diffuse or multifocal brain abnormalities. Additionally, the effect of surgical resection may wane over time.
Corpus Callosotomy
- Rationale: The corpus callosum is the main pathway for interhemispheric spread of epileptic activity. Severing connections between the hemispheres hampers the spread of ictal activity.
- Procedure: Anterior one-half to four-fifths callosotomy is typically performed, sparing the splenium to preserve some interhemispheric transfer of perceptual information and reduce complications of disconnection syndrome.
- Effectiveness: In one study, 38.8% of patients were free of disabling seizures one year after surgery, and 33.3% at two years. Significant and satisfactory seizure reduction (>85%) was achieved in about two-thirds of patients.
- Complications: Permanent serious complications are rare; most adverse effects are temporary, such as disconnection syndrome.
- Comparison: Total corpus callosotomy generally shows a higher response rate but also a higher chance of adverse effects compared to anterior corpus callosotomy.
Vagus Nerve Stimulation (VNS)
- Rationale: VNS is a palliative treatment for drug-resistant epilepsy in patients not candidates for resective brain surgery.
- Mechanism: The exact mechanism is unknown, but VNS may interrupt synchronous electrical activity characteristic of epileptic seizures and cause widespread changes in blood flow and metabolism in several cortical and subcortical regions.
- Effectiveness: In a study of 347 children, 32.5%, 37.6%, and 43.8% had ≥50% reduction in baseline seizure frequency at 6, 12, and 24 months post-implantation, respectively. In another study of 43 adults, 63% had ≥50% reduction in seizure frequency at 18 months post-implantation. Overall, about 50% of patients achieve ≥50% reduction in baseline seizure frequency.
- Adverse Effects: Common adverse effects include voice alteration, increased drooling, dyspnea, and coughing. Adverse effects are relatively minimal compared to corpus callosotomy.
Choosing Between Corpus Callosotomy and VNS
- Atonic Seizures: Corpus callosotomy might be preferred if atonic seizures predominate.
- Myoclonic Seizures: VNS might be preferred if myoclonic seizures prevail.
- Atypical Absence or Tonic-Clonic Seizures: Both procedures have similar effectiveness, but VNS might be preferred considering the adverse event profile.
- Meta-Analysis: Corpus callosotomy showed significantly better outcomes than VNS for >50% and >75% atonic seizure reduction. For all other seizure types, VNS offered comparable rates to corpus callosotomy.
Surgical interventions, including resective brain surgery, corpus callosotomy, and vagus nerve stimulation, provide additional treatment options for patients with drug-resistant LGS. Each approach has specific indications, benefits, and limitations, requiring careful consideration of the patient's clinical profile and seizure types.
Prognosis of Lennox-Gastaut Syndrome (LGS)
The long-term outcome for patients with Lennox-Gastaut Syndrome (LGS) is generally poor, and complete seizure freedom is rare. Epileptic encephalopathies in childhood, particularly LGS, are typically associated with significant long-term adverse effects on intellectual development, social functioning, and independent living. These unfavorable outcomes also have a substantial impact on family members and caregivers.
Long-Term Outcomes
- Seizure Control: Over time, patients with LGS may experience changes in seizure types and EEG characteristics. While the frequency and intensity of seizures tend to decrease with age, many patients continue to experience seizures, predominantly tonic or tonic-clonic seizures.
- Cognitive Function: In a retrospective study of 68 patients with LGS with a mean follow-up duration of 19.3 years, 94.7% of the patients had moderate to profound intellectual disability.
- Social and Independent Living: The long-term effects on intellectual and social functioning significantly impair the ability of patients to live independently.
Economic Impact
Lennox-Gastaut Syndrome is a costly disease. Patients with probable LGS have substantially higher healthcare costs compared to non-LGS patients, with total healthcare costs being 2-4 times higher. Medical costs are the primary drivers of these expenses. Timely diagnosis and appropriate treatment of LGS are likely to result in improved outcomes and potentially lower overall management costs.
The prognosis for LGS is generally unfavorable, with persistent seizures and significant cognitive impairment being common. The disease imposes considerable economic and emotional burdens on patients, families, and caregivers. Early diagnosis and effective treatment are crucial for improving patient outcomes and reducing the associated costs.
References
- Archer JS, Warren AE, Stagnitti MR, Masterton RA, Abbott DF, and Jackson GD (2014) Lennox-Gastaut syndrome and phenotype: secondary network epilepsies. Epilepsia 55(8):1245–1254
- Asadi-Pooya AA, Dlugos D, Skidmore C, Sperling MR (2017) Atlas of Electroencephalography, 3rd Edition. Epileptic Disord 19 (3):384. DOI: 10.1684/epd.2017.0934 PMID: 28872032.
- Camfield PR (2011) Definition and natural history of LennoxGastaut syndrome. Epilepsia 52(Suppl 5):3–9
- Camfield CS, Camfield PR, Gordon K, Wirrell E, Dooley JM (1996) Incidence of epilepsy in childhood and adolescence: a population based study in Nova Scotia from 1977 to 1985. Epilepsia 37:19–23
- Caraballo RH, Fortini S, Fresler S, Armeno M, Ariela A, Cresta A et al (2014) Ketogenic diet in patients with Lennox-Gastaut syndrome. Seizure 23(9):751–755
- Ferlazzo E, Nikanorova M, Italiano D, Bureau M, Dravet C, Calarese T et al (2010) Lennox-Gastaut syndrome in adulthood: clinical and EEG features. Epilepsy Res 89(2–3):271–277
- Lund C, Brodtkorb E, Rosby O, Rodningen OK, Selmer KK (2013) Copy number variants in adult patients with Lennox–Gastaut syndrome features. Epilepsy Res 105(1–2):110–117
- Pillay N, Archer JS, Badawy RA, Flanagan DF, Berkovic SF, Jackson G (2013) Networks underlying paroxysmal fast activity and slow spike and wave in Lennox-Gastaut syndrome. Neurology 81(7):665–673
- Trevathan E, Murphy CC, Yeargin-Allsopp M (1997) Prevalence and descriptive epidemiology of Lennox-Gastaut syndrome among Atlanta children. Epilepsia 38:1283–1288
- Warren AE, Abbott DF, Vaughan DN, Jackson GD, Archer JS (2016) Abnormal cognitive network interactions in LennoxGastaut syndrome: a potential mechanism of epileptic encephalopathy. Epilepsia 57(5):812–822
- Widdess-Walsh P, Dlugos D, Fahlstrom R, Joshi S, Shellhaas R, Boro A, EPGP Investigators et al (2013) Lennox-Gastaut syndrome of unknown cause: phenotypic characteristics of patients in the Epilepsy Phenome/Genome Project. Epilepsia 54(11):1898–1904
Principal source:
Asadi-Pooya AA (2018) Lennox-Gastaut syndrome: a comprehensive review. Neurol Sci 39 (3):403-414. DOI: 10.1007/s10072-017-3188-y PMID: 29124439.