Pulmonary function in Duchenne muscular dystrophy patients from the STRIDE Registry and CINRG Natural History Study: a matched cohort analysis
Már Tulinius, Filippo Buccella, Isabelle Desguerre, Janbernd Kirschner, Eugenio Mercuri, Francesco Muntoni, Andrés Nascimento Osorio, Shelley Johnson, Christian Werner, Joel Jiang, James Li, Panayiota Trifillis, Craig. M. McDonald
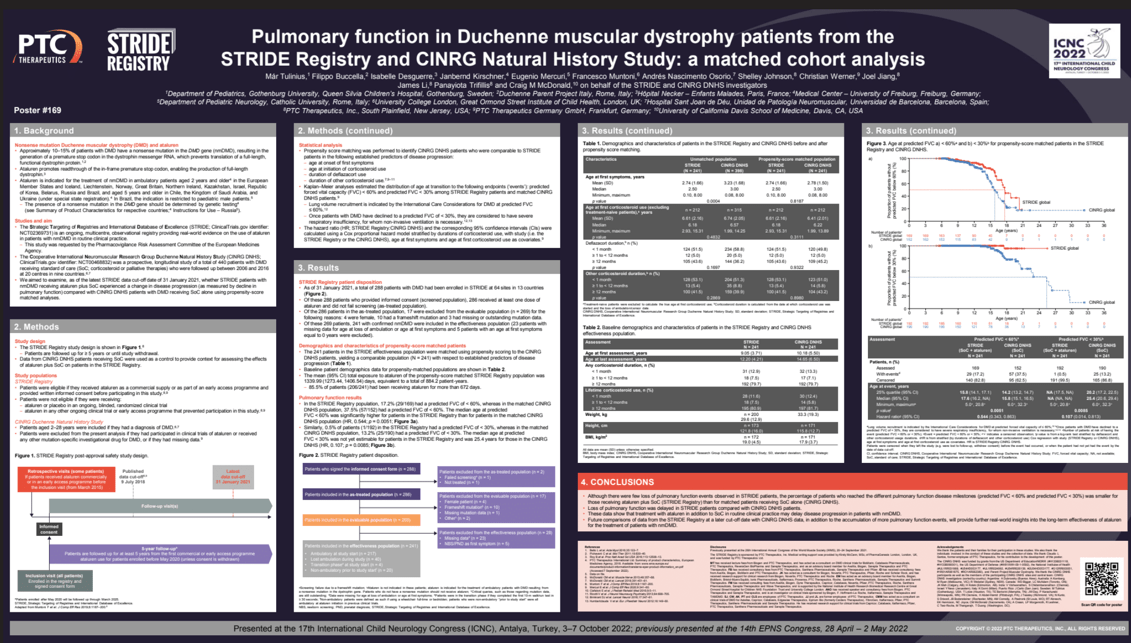
Objectives: We investigated if nonsense mutation Duchenne muscular dystrophy (nmDMD) patients receiving ataluren plus standard of care (SoC) in the STRIDE Registry (NCT02369731) experienced a lesser decline in pulmonary function versus DMD patients receiving SoC alone in the CINRG Duchenne Natural History Study (NCT00468832).
Methods: STRIDE is an ongoing, multicenter, observational registry providing data on ataluren use in nmDMD patients in routine clinical practice (data extracted, January 31, 2021). Propensity score matching identified STRIDE and CINRG patient cohorts (N=241) comparable in established predictors of disease progression: age at first symptoms, age at initiation of corticosteroid use, duration of deflazacort use, and duration of other corticosteroid use. CINRG patients who had received DMD investigational drugs were excluded. Kaplan–Meier analyses estimated ages at %-predicted forced vital capacity (FVC) <60% and <30%.
Results: Mean (SD) ages at onset of first symptoms (STRIDE vs CINRG; N=241 per cohort) were 2.7 (1.7) and 2.8 (1.5) years, respectively. Most patients (STRIDE vs CINRG) received corticosteroids for ≥12 months (79.7% per cohort); a similar proportion received deflazacort (43.6% vs 45.2%) or other corticosteroids (41.5% vs 43.2%). Median (95% CI) ages at %-predicted FVC <60% (STRIDE vs CINRG) were 17.6 (16.2, non-estimable) and 15.8 (15.1, 16.5) years, respectively (p=0.0051). Median (95% CI) ages at %-predicted FVC <30% were non-estimable for STRIDE patients (0.5% [1/192] of patients reached %-predicted FVC <30%) and 25.4 (20.6, 29.4) years for CINRG patients (p=0.0085).
Conclusion: These interim registry data suggest that ataluren plus SoC in routine clinical practice slows nmDMD disease progression.
Keywords: Duchenne muscular dystrophy; pulmonary function; STRIDE.
Már Tulinius
Gothenburg University, Queen Silvia Children’s Hospital
Sweden
Filippo Buccella
Parent Project APS
Italy
Isabelle Desguerre
Hôpital Necker – Enfants Malades
France
Janbernd Kirschner
Medical Center – University of Freiburg
Germany
Eugenio Mercuri
Catholic University
Italy
Francesco Muntoni
University College London, Great Ormond Street Institute of Child Health
United Kingdom
Andrés Nascimento Osorio
Hospital Sant Joan de Déu, Unidad de Patología Neuromuscular, Universidad de Barcelona
Spain
Shelley Johnson
PTC Therapeutics Inc.
United States
Christian Werner
PTC Therapeutics Germany GmbH
Germany
Joel Jiang
PTC Therapeutics Inc.
United States
James Li
PTC Therapeutics Inc.
Panayiota Trifillis
PTC Therapeutics Inc.
Craig. M. McDonald
University of California Davis School of Medicine
United States
Objectives: We investigated if nonsense mutation Duchenne muscular dystrophy (nmDMD) patients receiving ataluren plus standard of care (SoC) in the STRIDE Registry (NCT02369731) experienced a lesser decline in pulmonary function versus DMD patients receiving SoC alone in the CINRG Duchenne Natural History Study (NCT00468832).
Methods: STRIDE is an ongoing, multicenter, observational registry providing data on ataluren use in nmDMD patients in routine clinical practice (data extracted, January 31, 2021). Propensity score matching identified STRIDE and CINRG patient cohorts (N=241) comparable in established predictors of disease progression: age at first symptoms, age at initiation of corticosteroid use, duration of deflazacort use, and duration of other corticosteroid use. CINRG patients who had received DMD investigational drugs were excluded. Kaplan–Meier analyses estimated ages at %-predicted forced vital capacity (FVC) <60% and <30%.
Results: Mean (SD) ages at onset of first symptoms (STRIDE vs CINRG; N=241 per cohort) were 2.7 (1.7) and 2.8 (1.5) years, respectively. Most patients (STRIDE vs CINRG) received corticosteroids for ≥12 months (79.7% per cohort); a similar proportion received deflazacort (43.6% vs 45.2%) or other corticosteroids (41.5% vs 43.2%). Median (95% CI) ages at %-predicted FVC <60% (STRIDE vs CINRG) were 17.6 (16.2, non-estimable) and 15.8 (15.1, 16.5) years, respectively (p=0.0051). Median (95% CI) ages at %-predicted FVC <30% were non-estimable for STRIDE patients (0.5% [1/192] of patients reached %-predicted FVC <30%) and 25.4 (20.6, 29.4) years for CINRG patients (p=0.0085).
Conclusion: These interim registry data suggest that ataluren plus SoC in routine clinical practice slows nmDMD disease progression.
Keywords: Duchenne muscular dystrophy; pulmonary function; STRIDE.
Már Tulinius
Gothenburg University, Queen Silvia Children’s Hospital
Sweden
Filippo Buccella
Parent Project APS
Italy
Isabelle Desguerre
Hôpital Necker – Enfants Malades
France
Janbernd Kirschner
Medical Center – University of Freiburg
Germany
Eugenio Mercuri
Catholic University
Italy
Francesco Muntoni
University College London, Great Ormond Street Institute of Child Health
United Kingdom
Andrés Nascimento Osorio
Hospital Sant Joan de Déu, Unidad de Patología Neuromuscular, Universidad de Barcelona
Spain
Shelley Johnson
PTC Therapeutics Inc.
United States
Christian Werner
PTC Therapeutics Germany GmbH
Germany
Joel Jiang
PTC Therapeutics Inc.
United States
James Li
PTC Therapeutics Inc.
Panayiota Trifillis
PTC Therapeutics Inc.
Craig. M. McDonald
University of California Davis School of Medicine
United States