Comparison of North Star Ambulatory Assessment score change in nmDMD patients receiving ataluren: STRIDE Registry vs phase 3 clinical trial
Francesco Muntoni, Már Tulinius, Filippo Buccella, Isabelle Desguerre, Janbernd Kirschner, Andrés Nascimento Osorio, Shelley Johnson, Christian Werner, Joel Jiang, James Li, Panayiota Trifillis, Eugenio Mercuri
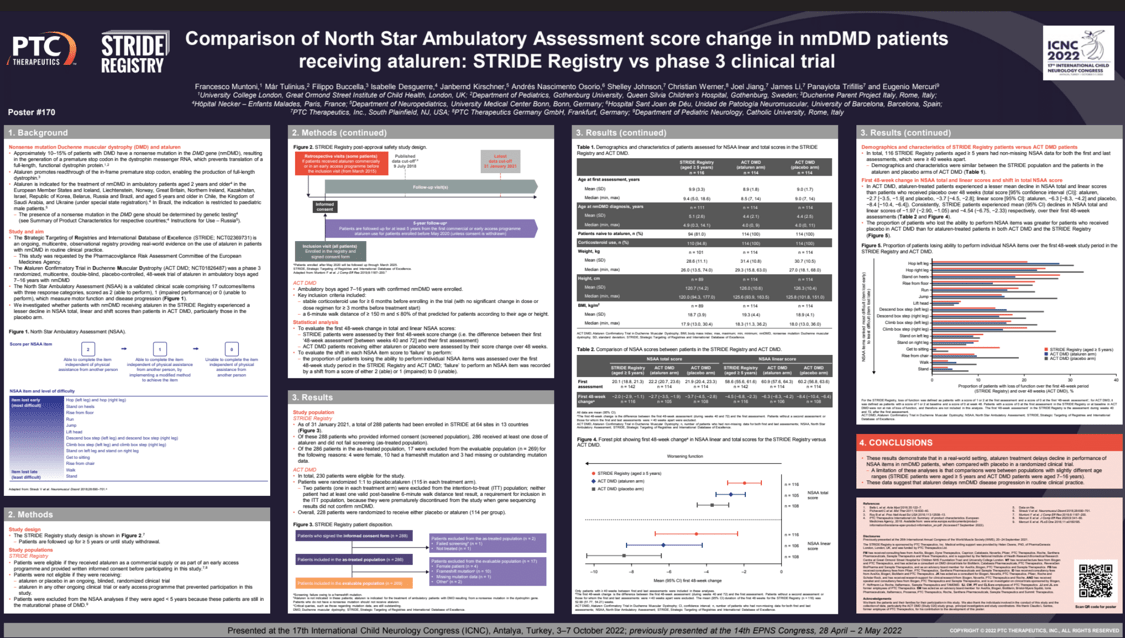
Objectives: We investigated whether nonsense mutation Duchenne muscular dystrophy (nmDMD) patients receiving ataluren in real-world practice (STRIDE Registry; NCT02369731) experienced lesser declines in North Star Ambulatory Assessment (NSAA) total, linear and shift scores vs patients receiving ataluren/placebo in a phase 3 clinical trial (Study 020; NCT01826487). The NSAA comprises 17 items, scored to document progressive loss of function.
Methods: STRIDE patients were assessed by first 48-week score change (difference between their first ‘48-week assessment’ [between 40 and 72 weeks] and first assessment); Study 020 patients were assessed by change over 48 weeks. The proportion of STRIDE patients who lost ability to perform NSAA items over the first 48 weeks was compared with Study 020 patients in a shift analysis; item failure was recorded by a shift from a score of 2 (able) or 1 (impaired) to 0 (unable).
Results: In Study 020, ataluren-treated patients experienced a lesser mean decline in NSAA total and linear scores vs placebo-allocated patients over 48 weeks (total score [95% confidence interval [CI]: ataluren,−2.7[−3.5,−1.9]; placebo,−3.7[−4.5,−2.8]; linear score [95% CI]: ataluren,−6.3[−8.3,−4.2]; placebo,−8.4[−10.4,−6.4]). STRIDE patients consistently experienced a mean (95% CI) decline in NSAA total and linear scores of −1.97(−2.90,−1.05) and −4.54(−6.75,−2.33) respectively, over their first 48-week assessments. The proportion of patients who lost ability to perform NSAA items was greater for Study 020 placebo-allocated patients than ataluren-treated STRIDE and Study 020 patients.
Conclusion: These results demonstrate that ataluren delays decline in performance of NSAA items in nmDMD patients vs placebo, indicating that ataluren delays disease progression.
Keywords: Duchenne muscular dystrophy; NSAA; STRIDE.
Francesco Muntoni
University College London, Great Ormond Street Institute of Child Health
United Kingdom
Már Tulinius
Gothenburg University, Queen Silvia Children’s Hospital
Sweden
Filippo Buccella
Parent Project APS
Italy
Isabelle Desguerre
Hôpital Necker – Enfants Malades
France
Janbernd Kirschner
Medical Center – University of Freiburg
Germany
Andrés Nascimento Osorio
Hospital Sant Joan de Déu, Unidad de Patología Neuromuscular, Universidad de Barcelona
Spain
Shelley Johnson
PTC Therapeutics Inc.
United States
Christian Werner
PTC Therapeutics Germany GmbH
Germany
Joel Jiang
PTC Therapeutics Inc.
United States
James Li
PTC Therapeutics Inc.
United States
Panayiota Trifillis
PTC Therapeutics Inc.
United States
Eugenio Mercuri
Catholic University
Italy
Objectives: We investigated whether nonsense mutation Duchenne muscular dystrophy (nmDMD) patients receiving ataluren in real-world practice (STRIDE Registry; NCT02369731) experienced lesser declines in North Star Ambulatory Assessment (NSAA) total, linear and shift scores vs patients receiving ataluren/placebo in a phase 3 clinical trial (Study 020; NCT01826487). The NSAA comprises 17 items, scored to document progressive loss of function.
Methods: STRIDE patients were assessed by first 48-week score change (difference between their first ‘48-week assessment’ [between 40 and 72 weeks] and first assessment); Study 020 patients were assessed by change over 48 weeks. The proportion of STRIDE patients who lost ability to perform NSAA items over the first 48 weeks was compared with Study 020 patients in a shift analysis; item failure was recorded by a shift from a score of 2 (able) or 1 (impaired) to 0 (unable).
Results: In Study 020, ataluren-treated patients experienced a lesser mean decline in NSAA total and linear scores vs placebo-allocated patients over 48 weeks (total score [95% confidence interval [CI]: ataluren,−2.7[−3.5,−1.9]; placebo,−3.7[−4.5,−2.8]; linear score [95% CI]: ataluren,−6.3[−8.3,−4.2]; placebo,−8.4[−10.4,−6.4]). STRIDE patients consistently experienced a mean (95% CI) decline in NSAA total and linear scores of −1.97(−2.90,−1.05) and −4.54(−6.75,−2.33) respectively, over their first 48-week assessments. The proportion of patients who lost ability to perform NSAA items was greater for Study 020 placebo-allocated patients than ataluren-treated STRIDE and Study 020 patients.
Conclusion: These results demonstrate that ataluren delays decline in performance of NSAA items in nmDMD patients vs placebo, indicating that ataluren delays disease progression.
Keywords: Duchenne muscular dystrophy; NSAA; STRIDE.
Francesco Muntoni
University College London, Great Ormond Street Institute of Child Health
United Kingdom
Már Tulinius
Gothenburg University, Queen Silvia Children’s Hospital
Sweden
Filippo Buccella
Parent Project APS
Italy
Isabelle Desguerre
Hôpital Necker – Enfants Malades
France
Janbernd Kirschner
Medical Center – University of Freiburg
Germany
Andrés Nascimento Osorio
Hospital Sant Joan de Déu, Unidad de Patología Neuromuscular, Universidad de Barcelona
Spain
Shelley Johnson
PTC Therapeutics Inc.
United States
Christian Werner
PTC Therapeutics Germany GmbH
Germany
Joel Jiang
PTC Therapeutics Inc.
United States
James Li
PTC Therapeutics Inc.
United States
Panayiota Trifillis
PTC Therapeutics Inc.
United States
Eugenio Mercuri
Catholic University
Italy